- 2017-09-22 22:23:42 整理: 李睿; 补充: 康文渊; 修订: 李继存
在本教程的第一部分中, 我们整理了GROMACS处理非标准残基的理论流程, 但如果你不熟悉GROMACS的力场结构, 单单根据那个流程来处理非标准残基仍然存在很大困难. 为此, 在本教程的第二部分中, 我们以一个具体实例来说明使用GROMACS时, 如何处理非标准残基, 如何在GROMACS力场文件中添加新的残基数据.
我们要处理的蛋白是GIC, 它是一种具有生理活性的芋螺毒素小肽, 其主要作用是抑制乙酰胆碱受体的开放, 因此有很大的潜力被开发成为一种止痛药. 在开发过程中, 有文章报道使用伽马氨基丁酸修饰后GIC会改变其活性. 这种修饰后的小肽被称为GID, 其中的伽马氨基丁酸残基CGU是一种非标准氨基酸残基. 因此, 如果想要使用GROMACS对GID进行分子动力学模拟, 就必须处理非标准残基CGU.
由于要模拟的体系是蛋白, 因此我们选用amber99sb-ildn力场. 但教程中的处理方法同样适用于其他力场.
第一步 获取蛋白的PDB文件
GID的PDB登录号为1MTQ, 下载其PDB文件1mtq.pdb.
下载后, 按常规步骤查看并清洗pdb文件, 去除杂原子, 去除重复模型, 只保留一个蛋白模型, 将其保存为gid.pdb. GID结构如下
查看pdb文件或通过可视化软件显示可知, GID中实际包含了两种非标准残基, CGU和HYP. 但GROMACS自带的amber99sb-ildn力场中包含了HYP残基的数据, 所以我们不需要对HYP使用非标准残基的处理流程, 只要稍加处理即可(具体处理见后文). 而对于CGU残基, 我们必须按照非标准残基的流程进行处理. 幸运的是, HYP残基为我们提供了一个非标准残基的示例, 如果在处理CGU时遇到问题(主要是准备其rtp文件), 可以参考amber99sb-ildn力场中HYP的数据.
第二步 更新残基类型文件residuetypes.dat
简单地讲, 对GROMACS而言, 所谓非标准残基就是指pdb2gmx不能识别的化学基团, 或者说是GROMACS的残基数据库中没有对应数据的化学基团. 为了使pdb2gmx能够识别新定义的残基, 并进而正确处理, 我们首先要将新残基的名称添加到/GMX安装路径/share/gromacs/top/residuetypes.dat文件中, 并指定新残基的类型. 浏览下此文件就可以看出, 残基的可能类型有Protein(蛋白), DNA, RNA, Water(水), Ion(离子). 我们要添加的CGU残基属于蛋白, 且处于肽链中间, 而不处于肽链两端, 所以只要简单地在residuetypes.dat文件中添加一行指明其类型是蛋白就可以了. 为简单起见, 我们将其添加到文件的最前面, 如下所示
| residuetypes.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 | CGU Protein ; 新添加的CGU残基
ABU Protein
ACE Protein
AIB Protein
ALA Protein
ARG Protein
ARGN Protein
ASN Protein
ASN1 Protein
ASP Protein
ASP1 Protein
...(下略)
|
这里多说几句, 如果你浏览一下residuetypes.dat文件的内容, 可能会发现
- 如果要添加的蛋白残基有可能处于端基位置, 还需要定义它处于N端和/或C端时的残基名称
- 如果要添加的残基属于DNA或RNA, 且有可能处于3端和/或5端, 同样需要定义其处于端基时的名称
- 如果要添加的残基可能存在不同的质子化状态, 同样需要定义其处于不同质子化状态时的名称
具体如何定义, 相信你能够从文件中得到答案.
第三步 创建包含残基的完整分子
为了将CGU残基的拓扑信息添加到GROMACS残基数据库中, 我们首先需要得到CGU的拓扑数据. 为此, 我们需要先将CGU残基从肽链中截取下来, 形成一个包含CGU的, 单独的, 完整的分子, 从而能够利用工具获得它的拓扑文件. 由于我们使用的是AMBER力场, 所以我们将使用ambertools获得这个分子的拓扑文件.
在抽取分子时, 如果我们只截取属于CGU的原子, 那么得到的并不是一个完整的分子, 需要进行封端处理. 常规的封端方法是使用氢原子. 但对于我们的情况并不合适, 因为如果只是对CGU用氢原子进行封端形成完整分子, 那么ambertools在判断CGU中每个原子的类型时, 很可能会与实际情况不符, 因为在实际的肽链中, 与CGU前后相连的是氨基酸残基的碳原子和氮原子. 所以最好是将CGU前后相邻的两个残基一起截取下来, 然后将两个相邻的残基用氢原子封端. 这样做时, 如果得到的分子过大, 导致ambertools不易处理, 也可以根据情况, 只截取相邻残基中与CGU的氮原子和碳原子最接近的一部分原子, 再使用氢原子封端. 有时候, 也可以分别使用乙酰基和甲胺基对截取残基的氮原子和碳原子进行封端. 这应该是能够使用的最小的封端基团了.
我们在这里将截取与CGU相邻的两个残基, 3ASP与5CYS, 然后用氢原子对它们封端. 所以, 我们截取gid.pdb中属于3ASP-4CGU-5CYS残基的原子. 截取原子时, 我们可以手动操作, 也可以使用PyMOL等可视化软件. 将截取的原子坐标保存为cgu.pdb. 其内容如下
| cgu.pdb | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 | ATOM 44 N ASP A 3 -9.246 3.307 -0.925 1.00 0.00 N
ATOM 45 CA ASP A 3 -8.653 1.995 -0.711 1.00 0.00 C
ATOM 46 C ASP A 3 -7.319 2.165 -0.007 1.00 0.00 C
ATOM 47 O ASP A 3 -6.489 2.980 -0.425 1.00 0.00 O
ATOM 48 CB ASP A 3 -8.442 1.253 -2.032 1.00 0.00 C
ATOM 49 CG ASP A 3 -7.601 0.001 -1.854 1.00 0.00 C
ATOM 50 OD1 ASP A 3 -8.029 -0.906 -1.107 1.00 0.00 O
ATOM 51 OD2 ASP A 3 -6.502 -0.065 -2.443 1.00 0.00 O
ATOM 52 H ASP A 3 -8.652 4.081 -0.978 1.00 0.00 H
ATOM 53 HA ASP A 3 -9.317 1.423 -0.080 1.00 0.00 H
ATOM 54 HB2 ASP A 3 -9.401 0.969 -2.438 1.00 0.00 H
ATOM 55 HB3 ASP A 3 -7.939 1.908 -2.731 1.00 0.00 H
HETATM 56 N CGU A 4 -7.117 1.414 1.061 1.00 0.00 N
HETATM 57 CA CGU A 4 -5.883 1.503 1.816 1.00 0.00 C
HETATM 58 C CGU A 4 -4.825 0.568 1.248 1.00 0.00 C
HETATM 59 O CGU A 4 -4.335 -0.330 1.933 1.00 0.00 O
HETATM 60 CB CGU A 4 -6.123 1.219 3.298 1.00 0.00 C
HETATM 61 CG CGU A 4 -5.865 2.430 4.184 1.00 0.00 C
HETATM 62 CD1 CGU A 4 -7.092 3.333 4.198 1.00 0.00 C
HETATM 63 CD2 CGU A 4 -4.516 3.045 3.826 1.00 0.00 C
HETATM 64 OE11 CGU A 4 -7.550 3.743 3.108 1.00 0.00 O
HETATM 65 OE12 CGU A 4 -7.605 3.624 5.295 1.00 0.00 O
HETATM 66 OE21 CGU A 4 -4.446 4.273 3.622 1.00 0.00 O
HETATM 67 OE22 CGU A 4 -3.525 2.287 3.734 1.00 0.00 O
HETATM 68 H CGU A 4 -7.815 0.792 1.350 1.00 0.00 H
HETATM 69 HA CGU A 4 -5.523 2.518 1.717 1.00 0.00 H
HETATM 70 HB2 CGU A 4 -5.467 0.423 3.612 1.00 0.00 H
HETATM 71 HB3 CGU A 4 -7.149 0.911 3.435 1.00 0.00 H
HETATM 72 HG CGU A 4 -5.762 2.055 5.191 1.00 0.00 H
ATOM 73 N CYS A 5 -4.475 0.798 -0.010 1.00 0.00 N
ATOM 74 CA CYS A 5 -3.472 0.008 -0.700 1.00 0.00 C
ATOM 75 C CYS A 5 -2.121 0.196 -0.002 1.00 0.00 C
ATOM 76 O CYS A 5 -1.236 -0.649 -0.079 1.00 0.00 O
ATOM 77 CB CYS A 5 -3.408 0.469 -2.164 1.00 0.00 C
ATOM 78 SG CYS A 5 -2.963 -0.811 -3.398 1.00 0.00 S
ATOM 79 H CYS A 5 -4.906 1.534 -0.497 1.00 0.00 H
ATOM 80 HA CYS A 5 -3.762 -1.032 -0.658 1.00 0.00 H
ATOM 81 HB2 CYS A 5 -4.376 0.853 -2.443 1.00 0.00 H
ATOM 82 HB3 CYS A 5 -2.686 1.266 -2.238 1.00 0.00 H
|
使用分子编辑软件(我使用的是GaussView)打开cgu.pdb, 检查下分子, 看是否需要补加缺失的原子(大多是氢原子).
可以看到, CGU残基的两个羧基上都没有氢原子, 需要根据模拟条件(如pH)决定是否添加(也就是决定其质子化状态). 我们在这里假定CGU的O22和O24都需要补加氢原子. 此外, 3ASP的N1和O8, 5CYS的C32和S35也都需要使用氢原子进行封端. 使用GaussView的加氢功能补加这些氢原子, 然后将分子保存为cgu_h.pdb. GaussView给出的原子坐标如下
| cgu_h.pdb | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 | REMARK 1 File created by GaussView 5.0.9
ATOM 1 N ASP A 3 -9.246 3.307 -0.925 N
ATOM 2 CA ASP A 3 -8.653 1.995 -0.711 C
ATOM 3 C ASP A 3 -7.319 2.165 -0.007 C
ATOM 4 O ASP A 3 -6.489 2.980 -0.425 O
ATOM 5 CB ASP A 3 -8.442 1.253 -2.032 C
ATOM 6 CG ASP A 3 -7.601 0.001 -1.854 C
ATOM 7 OD1 ASP A 3 -8.029 -0.906 -1.107 O
ATOM 8 OD2 ASP A 3 -6.502 -0.065 -2.443 O
ATOM 9 H ASP A 3 -8.662 4.116 -0.983 H
ATOM 10 HA ASP A 3 -9.317 1.423 -0.080 H
ATOM 11 HB2 ASP A 3 -9.401 0.969 -2.438 H
ATOM 12 HB3 ASP A 3 -7.939 1.908 -2.731 H
HETATM 13 N CGU A 4 -7.117 1.414 1.061 N
HETATM 14 CA CGU A 4 -5.883 1.503 1.816 C
HETATM 15 C CGU A 4 -4.825 0.568 1.248 C
HETATM 16 O CGU A 4 -4.335 -0.330 1.933 O
HETATM 17 CB CGU A 4 -6.123 1.219 3.298 C
HETATM 18 CG CGU A 4 -5.865 2.430 4.184 C
HETATM 19 CD1 CGU A 4 -7.092 3.333 4.198 C
HETATM 20 CD2 CGU A 4 -4.516 3.045 3.826 C
HETATM 21 OE11 CGU A 4 -7.550 3.743 3.108 O
HETATM 22 OE12 CGU A 4 -7.605 3.624 5.295 O
HETATM 23 OE21 CGU A 4 -4.446 4.273 3.622 O
HETATM 24 OE22 CGU A 4 -3.525 2.287 3.734 O
HETATM 25 H CGU A 4 -7.815 0.792 1.350 H
HETATM 26 HA CGU A 4 -5.523 2.518 1.717 H
HETATM 27 HB2 CGU A 4 -5.467 0.423 3.612 H
HETATM 28 HB3 CGU A 4 -7.149 0.911 3.435 H
HETATM 29 HG CGU A 4 -5.762 2.055 5.191 H
ATOM 30 N CYS A 5 -4.475 0.798 -0.010 N
ATOM 31 CA CYS A 5 -3.472 0.008 -0.700 C
ATOM 32 C CYS A 5 -2.121 0.196 -0.002 C
ATOM 33 O CYS A 5 -1.233 -0.690 -0.101 O
ATOM 34 CB CYS A 5 -3.408 0.469 -2.164 C
ATOM 35 SG CYS A 5 -2.963 -0.811 -3.398 S
ATOM 36 H CYS A 5 -4.906 1.534 -0.497 H
ATOM 37 HA CYS A 5 -3.762 -1.032 -0.658 H
ATOM 38 HB2 CYS A 5 -4.376 0.853 -2.443 H
ATOM 39 HB3 CYS A 5 -2.686 1.266 -2.238 H
TER 40 CYS A 5
HETATM 41 H 0 -6.912 3.794 5.937 H
HETATM 42 H 0 -2.719 2.805 3.797 H
HETATM 43 H 0 -10.238 3.399 -1.014 H
HETATM 44 H 0 -5.791 0.007 -1.802 H
HETATM 45 H 0 -1.932 1.081 0.569 H
HETATM 46 H 0 -1.659 -0.900 -3.484 H
|
为了后面能够方便地使用ambertools获得这个分子的拓扑文件, 并整理拓扑信息, 我们需要对文件进行一些修改:
- 删除
TER标识, 因为是单个分子 - 所有原子都使用
HETATM类型, 因为不将分子视为肽链 - 补加的氢原子移动到其所属残基后面: 41, 42放到4CGU后面, 43, 44放到3ASP后面, 45, 46放到5CYS后面
- 调整残基位置, 4CGU放在最前面, 然后是3ASP, 最后是5CYS, 这样在后面整理拓扑信息时更方便
- 修改CGU中的氢原子的名称. 因为使用
pdb2gmx自动加氢时, 对氢原子的名称有规定. 如果在一个原子上同时添加多个氢原子, 那么这些氢原子名称中的编号必须从1开始. 而CGU中HB2和HB3的编号不是从1开始, 所以要分别改为HB1和HB2. 同样, 补加的羧基氢原子分别更名为HE1和HE2. - 将3ASP的氮原子和碳原子名称后面添加
_, 5CYS的后面添加__. 这样可以区分不同残基, 方便后面整理拓扑信息. - 所有原子的残基名称统一改为
MOL, 链标识改为A, 残基编号为1. - 3ASP和5CYS的氢原子名称可以不用管, 因为最终我们只需要CGU的拓扑信息.
注意:
- GROMACS残基数据库中, 每个残基中各个原子的名称唯一且彼此间不能重复.
- 残基分子中与相邻氨基酸残基相连的原子名称必须为N和C, 否则形成拓扑文件时残基将无法与相邻残基形成正确的键.
将修改后的文件仍保存为cgu_H.pdb, 其内容如下:
| cgu_H.pdb | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 | HETATM 13 N MOL A 1 -7.117 1.414 1.061 N
HETATM 14 CA MOL A 1 -5.883 1.503 1.816 C
HETATM 15 C MOL A 1 -4.825 0.568 1.248 C
HETATM 16 O MOL A 1 -4.335 -0.330 1.933 O
HETATM 17 CB MOL A 1 -6.123 1.219 3.298 C
HETATM 18 CG MOL A 1 -5.865 2.430 4.184 C
HETATM 19 CD1 MOL A 1 -7.092 3.333 4.198 C
HETATM 20 CD2 MOL A 1 -4.516 3.045 3.826 C
HETATM 21 OE11 MOL A 1 -7.550 3.743 3.108 O
HETATM 22 OE12 MOL A 1 -7.605 3.624 5.295 O
HETATM 23 OE21 MOL A 1 -4.446 4.273 3.622 O
HETATM 24 OE22 MOL A 1 -3.525 2.287 3.734 O
HETATM 25 H MOL A 1 -7.815 0.792 1.350 H
HETATM 26 HA MOL A 1 -5.523 2.518 1.717 H
HETATM 27 HB1 MOL A 1 -5.467 0.423 3.612 H
HETATM 28 HB2 MOL A 1 -7.149 0.911 3.435 H
HETATM 29 HG MOL A 1 -5.762 2.055 5.191 H
HETATM 41 HE1 MOL A 1 -6.912 3.794 5.937 H
HETATM 42 HE2 MOL A 1 -2.719 2.805 3.797 H
HETATM 1 N_ MOL A 1 -9.246 3.307 -0.925 N
HETATM 2 CA MOL A 1 -8.653 1.995 -0.711 C
HETATM 3 C_ MOL A 1 -7.319 2.165 -0.007 C
HETATM 4 O MOL A 1 -6.489 2.980 -0.425 O
HETATM 5 CB MOL A 1 -8.442 1.253 -2.032 C
HETATM 6 CG MOL A 1 -7.601 0.001 -1.854 C
HETATM 7 OD1 MOL A 1 -8.029 -0.906 -1.107 O
HETATM 8 OD2 MOL A 1 -6.502 -0.065 -2.443 O
HETATM 9 H MOL A 1 -8.662 4.116 -0.983 H
HETATM 10 HA MOL A 1 -9.317 1.423 -0.080 H
HETATM 11 HB2 MOL A 1 -9.401 0.969 -2.438 H
HETATM 12 HB3 MOL A 1 -7.939 1.908 -2.731 H
HETATM 43 H MOL A 1 -10.238 3.399 -1.014 H
HETATM 44 H MOL A 1 -5.791 0.007 -1.802 H
HETATM 30 N__ MOL A 1 -4.475 0.798 -0.010 N
HETATM 31 CA MOL A 1 -3.472 0.008 -0.700 C
HETATM 32 C__ MOL A 1 -2.121 0.196 -0.002 C
HETATM 33 O MOL A 1 -1.233 -0.690 -0.101 O
HETATM 34 CB MOL A 1 -3.408 0.469 -2.164 C
HETATM 35 SG MOL A 1 -2.963 -0.811 -3.398 S
HETATM 36 H MOL A 1 -4.906 1.534 -0.497 H
HETATM 37 HA MOL A 1 -3.762 -1.032 -0.658 H
HETATM 38 HB2 MOL A 1 -4.376 0.853 -2.443 H
HETATM 39 HB3 MOL A 1 -2.686 1.266 -2.238 H
HETATM 45 H MOL A 1 -1.932 1.081 0.569 H
HETATM 46 H MOL A 1 -1.659 -0.900 -3.484 H
|
结构如下
第四步 获取残基的拓扑文件
使用与创建AMBER GAFF力场拓扑文件类似的方法获取残基分子的拓扑文件, 详细过程可参考 使用AmberTools+ACPYPE+Gaussian创建小分子GAFF力场的拓扑文件.
作为示例, 我们使用AM1-BCC电荷. 下面的脚本将上面博文中的几个步骤进行了整合, 方便使用.
| cgu.bsh | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | mol=cgu_h # pdb文件名称
# 1. 判断原子类型, 计算电荷.
# 注意我们要使用AMBER而不是GAFF的原子类型
# 我们没有指定体系的电荷和多重度, 因为它们与默认的电荷0, 多重度1一致
antechamber -i $mol.pdb -fi pdb -o $mol.mol2 -fo mol2 -c bcc -at amber -pf y
# 2. 检查补充缺失参数
parmchk2 -i $mol.mol2 -f mol2 -o $mol.mod
# 3. 生成AMBER参数文件和坐标文件
echo "
source leaprc.ff99SBildn
loadamberparams $mol.mod
mol=loadmol2 $mol.mol2
check mol
saveamberparm mol $mol.prm $mol.crd
quit" | sleap
# 4. 生成GROMACS拓扑文件和坐标文件
acpype -p $mol.prm -x $mol.crd -d
|
运行完成后, 注意检查AMBER优化后的构型sqm.pdb是否正常. 构型正常情况下后续步骤得到的结果才正确. 否则的话, 需要找出原因并修正错误.
完成本步骤后, 我们会得到残基分子的拓扑文件cgu_h_GMX.top.
我们优化后的构型如下, 看起来没有问题.
第五步 调整残基的电荷
我们需要明确, 对GROMACS残基数据库中的每个非离子状态的残基, 其净电荷应当为零, 这样它们组合成大分子后净电荷才正确. 但我们在上一步中计算出来的残基的净电荷, 除去用于封端的相邻残基后, 剩余部分的净电荷很可能不是零, 这就需要我们进行处理. 处理方法分两种情况:
- 如果采用RESP拟合电荷, 可以使用约束, 将每个残基的净电荷约束为零.
- 如果采用AM1-BCC电荷, 简单的处理方法是将相邻残基的净电荷加到相应的连接原子上, 或者将残基中所有原子的电荷减去其整体净电荷的其平均值.
这里我们使用调整连接原子电荷的方法. 所得拓扑文件中涉及电荷部分如下
| cgu_h_GMX.top | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 | [ moleculetype ]
;name nrexcl
cgu_h 3
[ atoms ]
; nr type resi res atom cgnr charge mass ; qtot bond_type
1 N 1 MOL N 1 -0.558901 14.01000 ; qtot -0.559
2 CT 1 MOL CA 2 0.038700 12.01000 ; qtot -0.520
3 C 1 MOL C 3 0.642101 12.01000 ; qtot 0.122
4 O 1 MOL O 4 -0.602101 16.00000 ; qtot -0.480
5 CT 1 MOL CB 5 -0.069400 12.01000 ; qtot -0.550
6 CT 1 MOL CG 6 -0.202700 12.01000 ; qtot -0.752
7 C 1 MOL CD1 7 0.641601 12.01000 ; qtot -0.111
8 C 1 MOL CD2 8 0.641601 12.01000 ; qtot 0.531
9 O 1 MOL OE11 9 -0.524501 16.00000 ; qtot 0.006
10 OH 1 MOL OE12 10 -0.579101 16.00000 ; qtot -0.573
11 O 1 MOL OE21 11 -0.524501 16.00000 ; qtot -1.097
12 OH 1 MOL OE22 12 -0.579101 16.00000 ; qtot -1.676
13 H 1 MOL H 13 0.362500 1.00800 ; qtot -1.314
14 H1 1 MOL HA 14 0.098700 1.00800 ; qtot -1.215
15 HC 1 MOL HB1 15 0.098200 1.00800 ; qtot -1.117
16 HC 1 MOL HB2 16 0.098200 1.00800 ; qtot -1.019
17 HC 1 MOL HG 17 0.121700 1.00800 ; qtot -0.897
18 HO 1 MOL HE1 18 0.460000 1.00800 ; qtot -0.437
19 HO 1 MOL HE2 19 0.460000 1.00800 ; qtot 0.023
; 以上为CGU
20 NT 1 MOL N_ 20 -0.900801 14.01000 ; qtot -0.878
21 CT 1 MOL C1 21 0.120500 12.01000 ; qtot -0.757
22 C 1 MOL C_ 22 0.660101 12.01000 ; qtot -0.097
23 O 1 MOL O1 23 -0.637101 16.00000 ; qtot -0.734
24 CT 1 MOL C3 24 -0.178400 12.01000 ; qtot -0.913
25 C 1 MOL C5 25 0.633101 12.01000 ; qtot -0.280
26 O 1 MOL OD1 26 -0.546001 16.00000 ; qtot -0.826
27 OH 1 MOL OD2 27 -0.604101 16.00000 ; qtot -1.430
28 H 1 MOL H1 28 0.370300 1.00800 ; qtot -1.059
29 H1 1 MOL H7 29 0.104700 1.00800 ; qtot -0.955
30 HC 1 MOL H9 30 0.094200 1.00800 ; qtot -0.861
31 HC 1 MOL HB3 31 0.094200 1.00800 ; qtot -0.766
32 H 1 MOL H2 32 0.370300 1.00800 ; qtot -0.396
33 HO 1 MOL H3 33 0.455000 1.00800 ; qtot 0.059
; 以上为3ASP
34 N 1 MOL N__ 34 -0.568901 14.01000 ; qtot -0.510
35 CT 1 MOL C2 35 -0.020300 12.01000 ; qtot -0.530
36 C 1 MOL C__ 36 0.554901 12.01000 ; qtot 0.025
37 O 1 MOL O2 37 -0.530101 16.00000 ; qtot -0.505
38 CT 1 MOL C4 38 -0.010300 12.01000 ; qtot -0.516
39 SH 1 MOL SG 39 -0.369900 32.06000 ; qtot -0.886
40 H 1 MOL H4 40 0.340500 1.00800 ; qtot -0.545
41 H1 1 MOL H8 41 0.124700 1.00800 ; qtot -0.420
42 H1 1 MOL H10 42 0.088200 1.00800 ; qtot -0.332
43 H1 1 MOL H11 43 0.088200 1.00800 ; qtot -0.244
44 HA 1 MOL H5 44 0.016200 1.00800 ; qtot -0.228
45 HS 1 MOL H6 45 0.227800 1.00800 ; qtot 0.000
; 以上为5CYS
|
检查拓扑文件可以看到, 实际上分子中每个残基的净电荷都很接近零. 其中CGU残基的净电荷为0.023, 很接近零, 不调整的话也影响不大, 但我们还是示例下如何调整.
调整电荷的方法是: 分别计算与CGU残基N端和C端相邻的残基的净电荷, 然后将其值分别累加到CGU残基的N原子和C原子上. 具体来说,
与CGU-N1相邻的是3ASP残基, 将其包含的每个原子的电荷累加, 得到其净电荷为0.0359980, 所以CGU-N1的电荷应当加上这个值, 调整为-0.558901+0.0359980=-0.522903. 同理, CGU-C3的电荷应调整为0.642101-0.0590010=0.5831. 这样调整之后CGU的净电荷就为零了.
第六步 整理残基的rtp条目
调整电荷后, 需要将拓扑文件整理为rtp格式, 这样才能将残基的拓扑数据添加到GROMACS力场的残基数据库中. rtp文件的详细说明见GROMACS中文手册第五章 5.6.1 残基数据库.
简单来说, rtp中需要定义的是残基中每个原子的名称, 类型和电荷, 以及原子间的成键相互作用项, 如原子间的连接信息, 键角信息, 二面角信息. 其实, 我们只要定义原子间的连接信息即可, 因为GROMACS可以根据原子间的连接信息自动推断出键角和二面角信息. 但有些特殊的成键相互作用, 如异常二面角, 与相邻残基形成的二面角仍然需要明确定义. 此外, 在定义这些成键相互作用项时可以忽略参数, 但考虑到ambertools给出的参数可能与GROMACS自带力场中的参数存在差异(特别是使用不同版本amber力场时), 再加上有些参数在GROMACS自带的力场中可能并不存在, 所以还是建议将ambertools给出的参数保留在rtp中.
首先将cgu_h_GMX.top另存为cgu.rtp.
删除不需要的内容, 只保留下列部分的内容: [ atoms ], [ bonds ], [ angles ], [ dihedrals ] ; propers, [ dihedrals ] ; impropers.
先整理[ atoms ]部分. 在rtp中, 这一部分只需要保留属于CGU残基的原子即可, 编号为1-19, 因为我们在前面的步骤中已经调整好顺序了, 此外也可以通过N_标识明显看出. rtp中只需要四列: 原子名称, 原子类型, 电荷, 编号. 这些信息在原来的拓扑文件中都是有的, 只不过顺序不同而已, 很容易使用列编辑功能整理好. 注意, CGU残基N1和C3原子的电荷应当是调整过的.
| cgu.rtp | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | [ atoms ]
; 名称 类型 电荷 编号
N N -0.522903 1
CA CT 0.038700 2
C C 0.5831 3
O O -0.602101 4
CB CT -0.069400 5
CG CT -0.202700 6
CD1 C 0.641601 7
CD2 C 0.641601 8
OE11 O -0.524501 9
OE12 OH -0.579101 10
OE21 O -0.524501 11
OE22 OH -0.579101 12
H H 0.362500 13
HA H1 0.098700 14
HB1 HC 0.098200 15
HB2 HC 0.098200 16
HG HC 0.121700 17
HE1 HO 0.460000 18
HE2 HO 0.460000 19
|
整理[ bonds ]部分. rtp文件中这一部分使用原子名称指定连接信息, 而不是像原拓扑文件中使用原子编号指定连接信息. 而我们得到的拓扑文件中最后的注释部分就是以原子名称指定的, 所以我们只要将这些内容提取出来, 稍加整理即可.
由于我们只需要保留与CGU残基有关的信息, 所以成键的两个原子中如果有任何一个编号大于19我们就将其删除. 根据原子编号的排列规律, 容易看出第二个的原子编号总是大于第一个, 所以我们直接删除第一个原子编号大于19的左右行即可.
将原拓扑文件中注释部分提取出来放到最前面, 删除原子名称中间的-字符, 同时删除标识势能函数类型的数字1.
最后, 将处于CGU前端并与其相邻的ASP残基的碳原子C_的名称改为-C, 指明是CGU的N原子与前一氨基酸残基的C原子相连. 值得注意的是, 并不需要为CGU残基末端的C原子定义C +N来指定与下一相邻残基的连接. 因为rtp中只定义每个残基的左边(N端)或者开始位置的连接, 末端连接是由下一个残基定义的. 所以删除C N__一行.
| cgu.rtp | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | [ bonds ]
N CA 1.4600e-01 2.7665e+05
N H 1.0090e-01 3.4326e+05
N -C 1.3450e-01 4.0016e+05
CA C 1.5080e-01 2.7472e+05
CA CB 1.5350e-01 2.5363e+05
CA HA 1.0930e-01 2.8108e+05
C O 1.2140e-01 5.4225e+05
CB CG 1.5350e-01 2.5363e+05
CB HB1 1.0920e-01 2.8225e+05
CB HB2 1.0920e-01 2.8225e+05
CG CD1 1.5080e-01 2.7472e+05
CG CD2 1.5080e-01 2.7472e+05
CG HG 1.0920e-01 2.8225e+05
CD1 OE11 1.2140e-01 5.4225e+05
CD1 OE12 1.3060e-01 3.9028e+05
CD2 OE21 1.2140e-01 5.4225e+05
CD2 OE22 1.3060e-01 3.9028e+05
OE12 HE1 9.7400e-02 3.0928e+05
OE22 HE2 9.7400e-02 3.0928e+05
|
对于[ angles ]和[ dihedrals ] ; propers部分, 处理方法类似[ bonds ]部分. 直接删除包含非CGU原子的项即可, 而且无需考虑与前后残基的连接原子. 有时我们甚至可以直接将这两部分删除, 但这样做有可能出现相互作用函数缺失的问题, 所以建议还是尽量保留.
| cgu.rtp | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 | [ angles ]
N CA C 1.1156e+02 5.5789e+02
N CA CB 1.1213e+02 5.5103e+02
N CA HA 1.0932e+02 4.1689e+02
CA N H 1.1678e+02 3.8526e+02
CA C O 1.2311e+02 5.6928e+02
CA CB CG 1.1063e+02 5.2894e+02
CA CB HB1 1.1005e+02 3.8802e+02
CA CB HB2 1.1005e+02 3.8802e+02
C CA CB 1.1053e+02 5.3379e+02
C CA HA 1.0766e+02 3.9857e+02
CB CA HA 1.1007e+02 3.8794e+02
CB CG CD1 1.1053e+02 5.3379e+02
CB CG CD2 1.1053e+02 5.3379e+02
CB CG HG 1.1005e+02 3.8802e+02
CG CB HB1 1.1005e+02 3.8802e+02
CG CB HB2 1.1005e+02 3.8802e+02
CG CD1 OE11 1.2311e+02 5.6928e+02
CG CD1 OE12 1.1220e+02 5.8442e+02
CG CD2 OE21 1.2311e+02 5.6928e+02
CG CD2 OE22 1.1220e+02 5.8442e+02
CD1 CG CD2 1.1161e+02 5.3605e+02
CD1 CG HG 1.0968e+02 3.9497e+02
CD1 OE12 HE1 1.0737e+02 4.2836e+02
CD2 CG HG 1.0968e+02 3.9497e+02
CD2 OE22 HE2 1.0737e+02 4.2836e+02
OE11 CD1 OE12 1.2288e+02 6.4752e+02
OE21 CD2 OE22 1.2288e+02 6.4752e+02
HB1 CB HB2 1.0835e+02 3.2995e+02
[ dihedrals ] ; propers
N CA C O 180.00 0.00000 2
N CA CB CG 0.00 0.65084 3
N CA CB HB1 0.00 0.65084 3
N CA CB HB2 0.00 0.65084 3
CA CB CG CD1 0.00 0.65084 3
CA CB CG CD2 0.00 0.65084 3
CA CB CG HG 0.00 0.66944 3
C CA CB CG 0.00 0.65084 3
C CA CB HB1 0.00 0.65084 3
C CA CB HB2 0.00 0.65084 3
CB CA C O 180.00 0.00000 2
CB CG CD1 OE11 180.00 0.00000 2
CB CG CD1 OE12 180.00 0.00000 2
CB CG CD2 OE21 180.00 0.00000 2
CB CG CD2 OE22 180.00 0.00000 2
CG CD1 OE12 HE1 180.00 9.62320 2
CG CD2 OE22 HE2 180.00 9.62320 2
CD1 CG CD2 OE21 180.00 0.00000 2
CD1 CG CD2 OE22 180.00 0.00000 2
CD2 CG CD1 OE11 180.00 0.00000 2
CD2 CG CD1 OE12 180.00 0.00000 2
OE11 CD1 OE12 HE1 0.00 7.94960 1
OE11 CD1 OE12 HE1 180.00 9.62320 2
OE21 CD2 OE22 HE2 0.00 7.94960 1
OE21 CD2 OE22 HE2 180.00 9.62320 2
H N CA C 0.00 0.00000 0
H N CA CB 0.00 0.00000 0
H N CA HA 0.00 0.00000 0
HA CA C O 0.00 0.00000 0
HA CA C O 0.00 3.34720 1
HA CA C O 180.00 0.33472 3
HA CA CB CG 0.00 0.65084 3
HA CA CB HB1 0.00 0.65084 3
HA CA CB HB2 0.00 0.65084 3
HB1 CB CG CD1 0.00 0.65084 3
HB1 CB CG CD2 0.00 0.65084 3
HB1 CB CG HG 0.00 0.62760 3
HB2 CB CG CD1 0.00 0.65084 3
HB2 CB CG CD2 0.00 0.65084 3
HB2 CB CG HG 0.00 0.62760 3
HG CG CD1 OE11 0.00 0.00000 0
HG CG CD1 OE11 0.00 3.34720 1
HG CG CD1 OE11 180.00 0.33472 3
HG CG CD1 OE12 180.00 0.00000 2
HG CG CD2 OE21 0.00 0.00000 0
HG CG CD2 OE21 0.00 3.34720 1
HG CG CD2 OE21 180.00 0.33472 3
HG CG CD2 OE22 180.00 0.00000 2
|
整理[ impropers ]部分. 首先将[ dihedrals ] ; impropers修改为[impropers]
, 然后删除存在编号大于19的项, 但保留C_- CA- N- H和CA- N__- C- O对应的项. 这样共保留4项, 其中两项对应于两个羧基上的氢原子, 剩下两项对应于CGU与前后残基所成肽键的异常二面角项. 同样, 我们需要将后面两项中的C_改为-C, N__改为+N指明是前后残基的相连原子, 而不是CGU自身的原子.
| ahk | |
|---|---|
1 2 3 4 5 | [ impropers ]
CA +N C O 180.00 4.60240 2
CG OE11 CD1 OE12 180.00 4.60240 2
CG OE21 CD2 OE22 180.00 4.60240 2
-C CA N H 180.00 4.60240 2
|
成键部分整理完成后, 在文件的开头添加默认成键类型, CGU残基的定义, 最终得到了满足要求的rtp文件, 其整个结构如下
| cgu.rtp | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 | [ bondedtypes ]
1 1 9 4 1 3 1 0
[ CGU ]
[ atoms ]
...
[ bonds ]
...
[ angles ]
...
[ dihedrals ] ; propers
...
[ impropers ]
...
|
第七步 准备残基的hdb文件
我们在使用pdb2gmx的时候, 经常需要使用-ignh选项来自动添加氢原子, 所以, 我们也需要为新残基CGU准备一个hdb文件, 用于说明如何自动添加氢原子. GROMACS手册第五章 5.6.4 氢数据库对此有说明, 但不够直观. 下面给出氢原子添加方式的图形化说明.
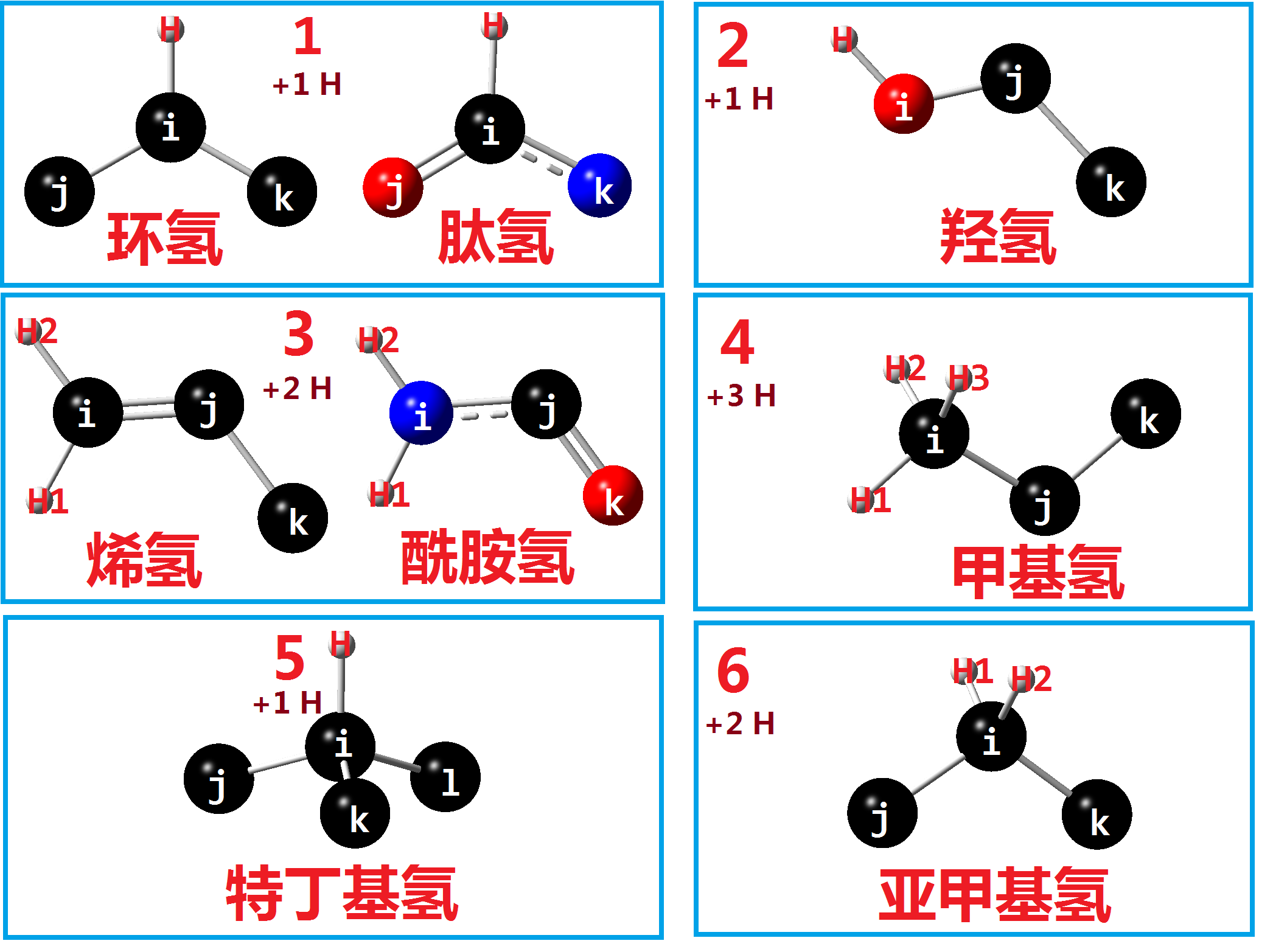
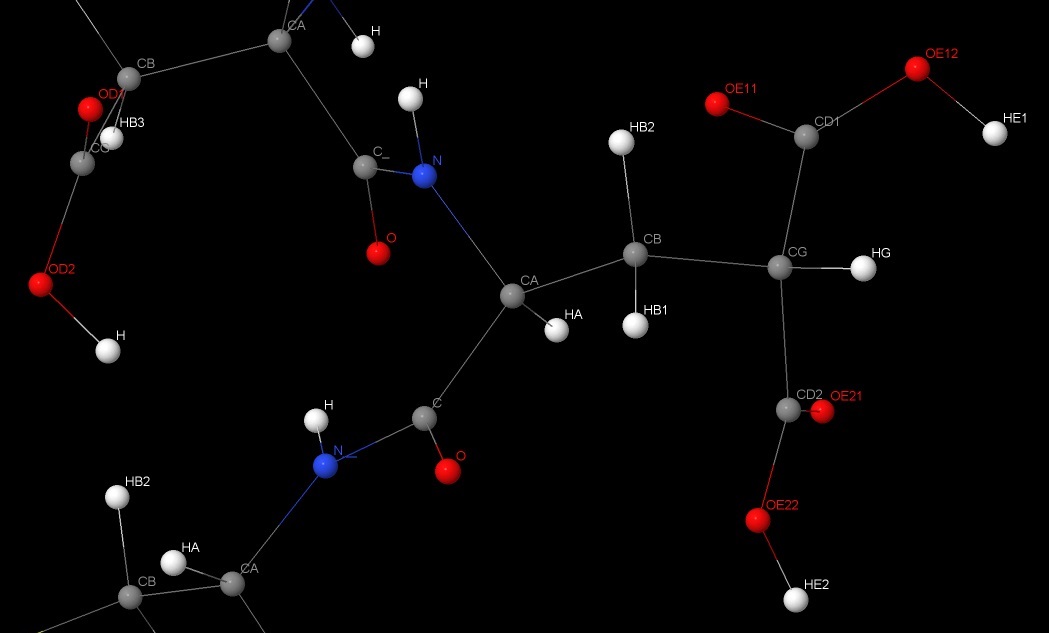
创建cgu.hdb文件, 根据CGU中氢原子的名称
定义hdb文件如下:
| cgu.hdb | |
|---|---|
1 2 3 4 5 6 7 | CGU 6
1 1 H N CA -C
1 5 HA CA N C CB
2 6 HB CB CA CG
1 5 HG CG CB CD1 CD2
1 2 HE1 OE12 CD1 CG
1 2 HE2 OE22 CD2 CG
|
其中第一行为残基名称, 待添加氢的类型数, 接下来每行列出一类待添加氢的信息: 加氢数目, 加氢方式, 加氢名称, 原子i, 原子j, 原子k, 原子l.
第八步 模拟测试
我们使用的是amber99sb-ildn力场, 所以将准备好的cgu.rtp和cgu.hdb文件放到
/GMX安装路径/share/gromacs/top/amber99sb-ildn.ff目录下, 这样CGU残基的信息就被添加到力场数据库中了. 这里要说明的是, 我们不建议直接将rtp和hdb里面的信息添加到GROMACS力场自带的aminoacids.rtp和aminoacids.hdb文件中, 那样做容易污染原先的文件.
需要注意的是, 由于原始gid.pdb中也含有HYP残基, 虽然我们使用的amber99sb-ildn力场中含有它的拓扑数据, 但因为它不是常见的标准残基, 所以我们要查看一下它的原子名称是否与残基数据库中的一致. 打开/GMX安装路径/share/gromacs/top/amber99sb-ildn.ff/aminoacids.rtp, 搜索HYP就可以看到HYP残基中每个原子名称, 与gid.pdb中的名称进行对比, 可以发现, gid.pdb中HYP的CD必须改为CD2才与力场数据库一致.
新残基添加到数据库中以后, 我们就可以使用pdb2gmx来测试是否添加成功了. 运行
gmx pdb2gmx -f gid.pdb -ignh
选择amber99sb-ildn力场, TIP3P水模型. 如果不能通过, 仔细检查cgu.rtp, 修正错误, 直至成功通过, 得到conf.gro和topol.top文件.
使用可视化软件打开conf.gro文件, 检查自动添加的氢原子是否正确. 如果不正确, 需要修改cgu.hdb, 重复上面的pdb2gmx命令, 直至添加的氢原子正确为止.
按照蛋白模拟的常规流程进行短时间的模拟, 检查轨迹是否有明显的异常(如原子跳跃异常, 成键断开等). 如果有明显异常, 寻找产生问题的原因. 修正后, 重复模拟, 直至轨迹正常.

其他可能的问题
-
我们在处理过程中, 使用了amber的原子类型, 目的在于尽量避免引入新的原子类型. 如果确实需要引入新的原子类型, 我们需要添加新的原子类型及其参数到力场文件中, 还需要定义新原子类型与其他原子类型的成键相互作用参数. 如果混用不同的力场, 很多成键参数是没有的, 处理起来很麻烦. 因此建议尽量避免引入新的原子类型, 不要混用力场.
-
如果
pdb2gmx执行成功, 而grompp时出错, 提示存在未定义的成键相互作用, 那么必须定义这些缺失的成键相互作用参数. 这些参数或者来自文献, 或者自行拟合, 或者使用力场中的类似参数. 通常少数几个成键参数的不同对结构和分子间相互作用的影响不大. 但情况并非总是这样, 所以你还是要留意这一点.
