- 原始文档: Benjamin D. Madej & Ross Walker, An Introduction to Molecular Dynamics Simulations using AMBER
- 更新至AMBER 15: Aditi Munshi and Ross Walker
- 2018-01-16 10:37:52 翻译: 刘仲奇; 校订: 李继存
本教程是专为那些完全没有运行过分子动力学模拟的新用户, 或只有少量模拟经验的用户准备的. 完成本教程不需要提前了解AMBER或Linux的知识, 但需要提前安装好AmberTools和VMD, 并正确设置AMBERHOME环境变量. 如果你是AMBER的新用户, 或对一般的MD毫无了解, 可通过此教程入门.
AMBER基础教程B0: AMBER分子动力学模拟入门
- 原始文档: Benjamin D. Madej & Ross Walker, An Introduction to Molecular Dynamics Simulations using AMBER
- 更新至AMBER 15: Aditi Munshi and Ross Walker
- 2018-01-16 10:37:52 翻译: 刘仲奇; 校订: 李继存
- AMBER基础教程B0: AMBER分子动力学模拟入门
- 介绍
- 开始使用Linux
- 准备拓扑文件和坐标文件
- 准备Amber MD的
sander输入文件 - 成品模拟输入
- 运行Amber MD模拟程序
sander- 17. 首先从终端将目录切换到
Tutorial目录, 其中存放了所有的输入文件.~是主目录的快捷方式, 其中有你创建的Tutorial目录. - 运行最小化
- 18. 用
sander运行丙氨酸二肽的最小化 - 最小化输出文件
- 19. 使用gedit打开输出文件
01_Min.out - 运行升温MD
- 20. 使用
sander升温丙氨酸二肽. - 加热输出文件
- 21. 打开输出文件
02_Heat.out查看系统输出. - 运行成品MD
- 22. 使用
sander运行丙氨酸二肽的成品MD - 23. 要监控作业的状态, 请使用程序
tail将输出文件显示到终端 - 24. 要退出
tail, 按<CTRL-C>退出程序. - 完成MD模拟
- 成品模拟输出
- 25. 用gedit打开输出文件
03_Prod.out, 查看MD模拟的输出.
- 17. 首先从终端将目录切换到
- 可视化结果
- 26. 要启动VMD, 请打开一个终端, 将目录更改为
Tutorial目录, 然后运行vmd. - 27. 使用
File -> New Molecule...创建一个新的分子 - 28. 为
New Molecule加载文件. 然后选择Amber参数和拓扑文件prmtop. 将文件类型设置为AMBER7.Parm. 点击Load. - 29. 为
0: prmtop加载文件. 然后选择Amber轨迹文件03_Prod.mdcrd. 将文件类型设置为AMBER Coordinates with Periodic Box. 点击Load. - 30. 在
Selected Atoms中输入all not water - 31. 在
Drawing Method中选择Licorice - 更多VMD信息
- 26. 要启动VMD, 请打开一个终端, 将目录更改为
- 分析MD结果
- 使用
cpptraj分析RMSD - 结论
- 附录: 文件
介绍
这个教程旨在介绍如何使用Amber进行分子动力学模拟. 它是围绕AMBER Tools v14设计的, 并假设您以前没有使用过Linux或者Amber. 它专门为想要了解如何运行分子动力学模拟的新用户而设计. 如果您的电脑上已经正确安装了Amber Tools v15, VMD和xmgrace就可以学习本教程.
AMBER 代表辅助的模型构建和能量精化(Assisted Model Building and Energy Refinement), 它不仅指代分子动力学程序, 也指代一组力场, 描述了生物分子相互作用的势能函数和参数.
为了在Amber中运行分子动力学模拟, 每个分子的相互作用都由分子力场描述. 力场为每个分子定义了特定的参数.
sander 是Amber中进行分子动力学的基本程序, pmemd 是一个高性能的MD程序, 包含了sander的一部分功能, 它还可以使用图形处理单元(GPU, graphics processing units)加速运行.
为了使用sander或者pmemd运行分子动力学模拟, 需要三个必备的文件:
prmtop: 描述系统中分子的参数和拓扑inpcrd: 描述系统初始分子坐标mdin: 描述Amber分子动力学程序的设置
开始使用Linux
Amber MD是一个完全基于命令行界面(CLI, Command Line Interface)的软件, 运行于Linux计算机上. 要运行Amber, 你需要打开一个终端.
1. 在你的Linux计算机上打开一个终端
在大多数Linux机器上, 您的终端类似下图:
本教程的大部分工作将主要通过终端完成.
列出文件并建立一个目录(文件夹)来储存你的文件
2. 使用ls命令列出当前目录中的内容
当你第一次登录并启动终端时, 您当前的工作目录(或文件夹)就是您的主目录. 它的名称与您的用户名相同, 并且这是您的文件和目录的存储位置. 在大多数情况下, 它的位置在/home/username. 我们可以使用ls(list)命令查看目录:
ls
此时, 您的主目录中可能会有一些文件和目录, 这些文件和目录是使用您的账户自动创建的.
3. 使用mkdir命令创建一个名为Tutorial的目录.
您将需要一个新的目录来存放本教程中创建的文件和文件夹. 所用的命令为mkdir(make directory).
mkdir Tutorial
现在输入ls命令, 你可以看到您的目录已经创建.
ls
显示
Tutorial
4. 使用cd命令切换不同的目录
现在, 您可能想要进入您的Tutorial目录, 这样您的工作文件都将被保存在这里. 这可以通过cd(change directory)命令完成.
cd Tutorial
ls
有一个特殊的目录, 名称为... 这是当前目录的父目录. 所以要想返回到Tutorial目录的父目录, 可使用cd ..:
cd ..
ls
显示
Tutorial
如果您需要返回到您的主目录, 请使用cd命令本身. 波浪号~是您的主目录的快捷方式. 以下的命令都可以将目录更改为您的主目录.
cd
cd ~
5. 使用pwd输出主目录的工作目录路径名称
路径名称描述了您所处的目录相对于整个计算机文件系统的位置. 您的主目录在整个文件系统中的位置看通过pwd(print working directory)命令获知:
cd ~
pwd
显示
/home/username
这是您所在的当前工作目录. 在这种情况下, 目录usename位于/(root)目录中的home目录中.
准备拓扑文件和坐标文件
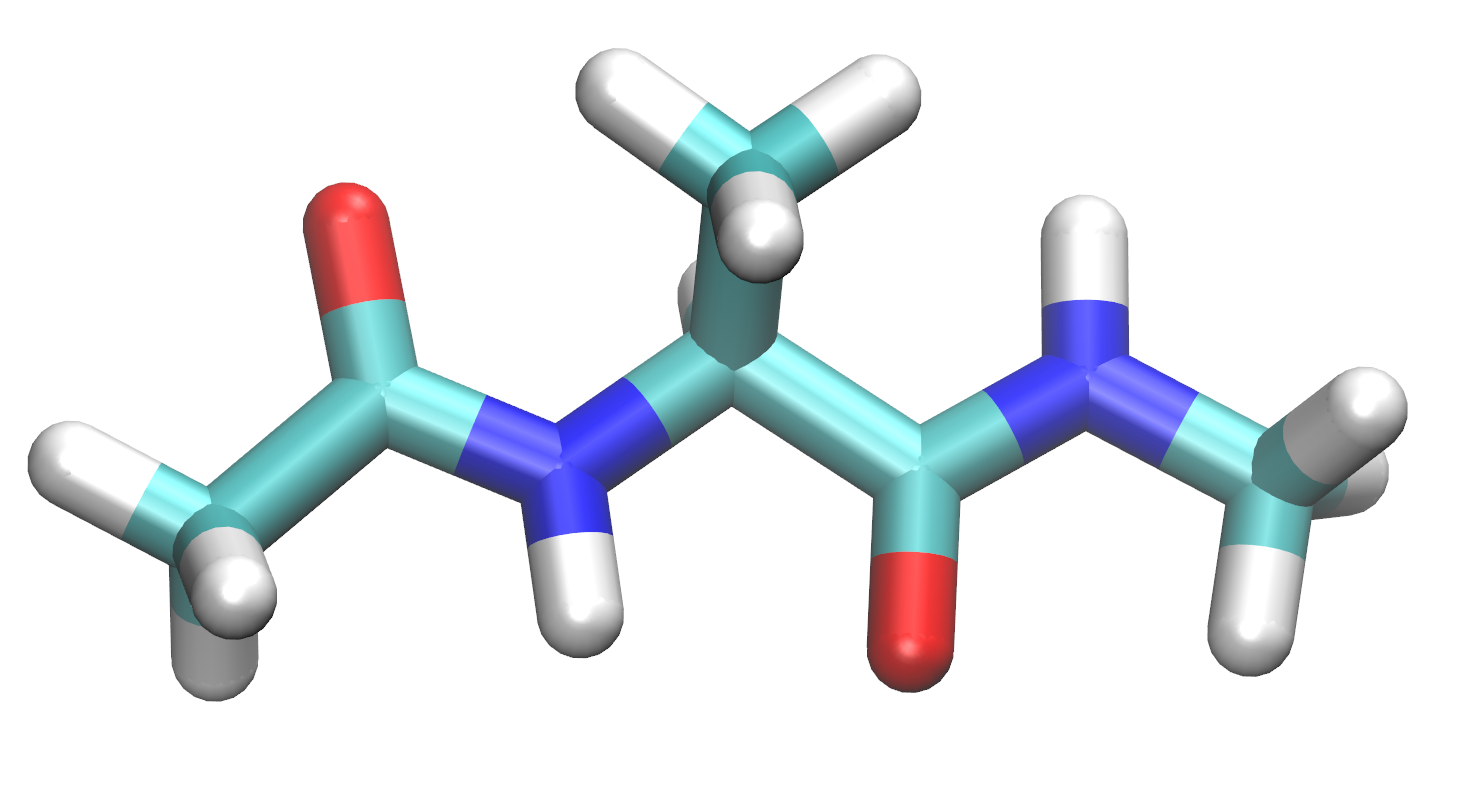
在本教程中, 您将在名为xLEaP的准备程序中构建以下分子以用于在AMBER中进行模拟.
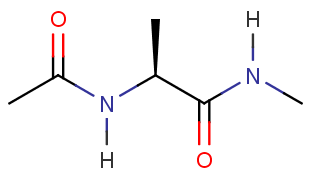
为了建立并溶剂化这个分子, 您需要启动xLEaP. xLEaP具有另一个命令行界面和简单的分子图形界面, 用于构建系统拓扑并为分子定义参数.
6. 使用xleap命令启动xLEaP
xleap
-I: Adding /usr/local/amber_14/amber/dat/leap/prep to search path.
-I: Adding /usr/local/amber_14/amber/dat/leap/lib to search path.
-I: Adding /usr/local/amber_14/amber/dat/leap/parm to search path.
-I: Adding /usr/local/amber_14/amber/dat/leap/cmd to search path.
您应该看到一个类似这样的窗口:
警告: 不要 在任何xLEaP窗口上点击X. 它将完全退出xLEaP.
注意: 这个时候, 关闭键盘的Num Lock以使菜单正常工作是个好主意.
加载蛋白质和核酸的力场
MD力场是由哈密顿量(势能函数)及其相关参数定义的, 它描述了系统中分子之间的分子内和分子间相互作用. 在MD中, 会积分哈密顿量以获得分子的力和速度.
Amber的哈密顿量的基本形式是:
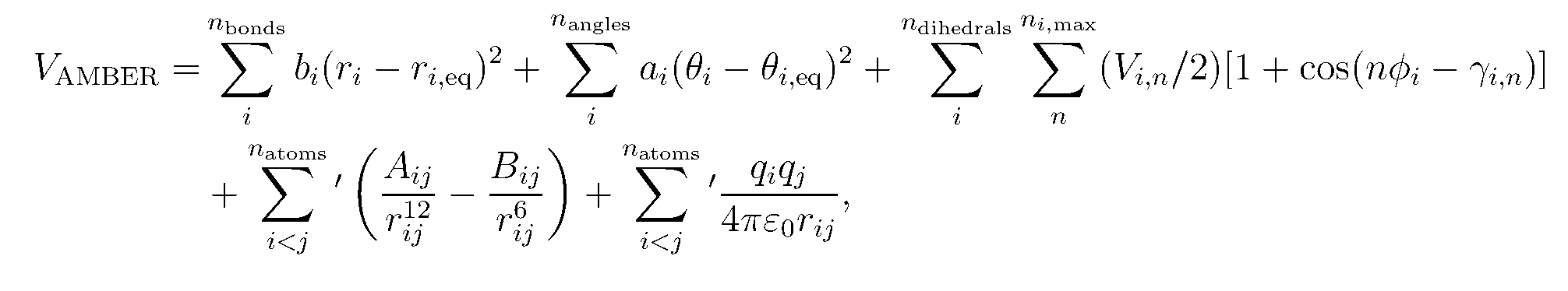
为了运行分子动力学模拟, 我们需要加载力场来描述丙氨酸二肽的势能. 对蛋白质和核酸我们将使用AMBER的FF14SB力场, FF14SB基于FF12SB, FF12SB是FF99SB的更新版本, 而FF99SB力场又是基于原始的Amber的Cornell等人(1995)的[ff94]力场. FF14SB力场最显著的变化包括更新了蛋白质Phi-Psi的扭转项, 并重新拟合了侧链的扭转项. 这些变化一起改进了对这些分子中α螺旋的估计.
7. 现在用source命令加载FF14SB力场
source leaprc.ff14SB
显示
Loading parameters:
/usr/local/amber14/dat/leap/parm/frcmod.ff14SB
Reading force field modification type file (frcmod)
Reading title:
建立丙氨酸二肽
我们可以将丙氨酸氨基酸的N端使用乙酰基封端, C端使用N-甲基酰胺封端, 这样就可以构建出丙氨酸二肽. 当加载了FF14SB力场之后, 就可以使用xLEaP中的这些”构建组件”来构建成分子. sequence(序列)命令可以利用已有组件创建一个新的组件并将它们连接在一起.
8. 使用sequence命令从ACE, ALA和NME组件创建一个名为foo的新组件
foo = sequence { ACE ALA NME }
现在得到了一个单独的丙氨酸二肽分子, 储存在组件foo. xLEaP提供了一个非常基本的编辑器来检查和更改组件和分子.
9. 检查丙氨酸二肽分子的结构, 使用edit命令来查看结构.
edit foo
编辑窗口将如下所示:
从这里, 您可以检查分子的拓扑, 结构, 原子名称, 原子类型和部分电荷. 也可以对分子进行基本的编辑.
警告: 不要点击X来关闭这个窗口, 这将完全退出xLEaP. 要关闭此窗口, 请使用Unit -> Close.
溶剂化丙氨酸二肽
准备丙氨酸二肽系统的下一步是用显式的水分子溶剂化分子. 在这个模拟中, 我们将添加TIP3P水分子到系统中.
在这种类型的模拟中, 系统具有周期性的边界条件, 这意味着离开系统一侧的分子将被转回到系统的另一侧. 周期性盒子足够大非常重要, 即丙氨酸二肽周围有足够的水, 以使丙氨酸二肽分子不与其自身的周期性映像相互作用.
有许多水模型可用于MD模拟. 但是, 对于本教程, 我们将使用TIP3P水模型进行模拟.
10. 使用solvatebox命令对系统进行溶剂化.
source leaprc.water.tip3p
solvatebox foo TIP3PBOX 10.0
TIP3PBOX指定要溶剂化的水盒子的类型. 10.0表示分子在丙氨酸二肽和周期性盒壁之间应该具有至少10 埃的缓冲区.
保存Amberprmtop和inpcrd输入文件
现在我们将保存prmtop和inpcrd文件到当前工作目录. 现在组件foo包括丙氨酸二肽分子, 水分子和模拟所需的周期性盒子信息. 参数将根据ff99SB力场指定.
11. 要保存prmtop和inpcrd文件, 请使用saveamberparm命令
saveamberparm foo prmtop inpcrd
显示
Checking Unit.
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
Building improper torsion parameters.
total 4 improper torsions applied
Building H-Bond parameters.
Incorporating Non-Bonded adjustments.
Not Marking per-residue atom chain types.
Marking per-residue atom chain types.
(Residues lacking connect0/connect1 -
these don't have chain types marked:
res total affected
WAT 630
)
(no restraints)
警告: 请仔细阅读此命令的输出, 特别是其中的警告和错误信息, 它们可能导致您的prmtop和inpcrd文件无法正确构建.
Amber参数/拓扑和坐标文件
丙氨酸二肽的prmtop和inpcrd文件可在这里下载: prmtop, inpcrd
退出xLEaP
12. 要退出xLEaP, 请使用quit.
quit
准备Amber MD的sander输入文件
所需的最后一个文件用于控制MD运行的输入文件, 其中定义了MD运行时的程序设置. 在本教程中, 我们将对系统进行能量最小化, 然后缓慢升温, 最后在所需的温度和压力下进行成品MD.
- 最小化
- 在恒定的体积和温度(NVT)下升温20 ps, 从0 K升温到300 K.
- 在1 atm和300 K的恒定压力和温度(NPT)下运行60 ps的成品MD.
我们每2 ps保存一次轨迹并写入输出文件一次, 使用Langevin恒温器控制温度, 使用随机种子初始化随机数发生器.
要控制所有这些设置, 我们将使用文本编辑器编写一个简单的输入文件. Linux下有许多可用的文本编辑器, 但我们将使用您的Linux计算机上自带的简单文本编辑器.
13. 在您的Linux计算机上打开gedit Text Editor.
gedit的界面如下所示:
最小化输入
14. 创建包含以下最小化设置的文件01_Min.in:
| 01_Min.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 | Minimize
&cntrl
imin=1,
ntx=1,
irest=0,
maxcyc=2000,
ncyc=1000,
ntpr=100,
ntwx=0,
cut=8.0,
/
|
这些设置总结如下:
imin=1: 选择运行能量最小化ntx=1: 从ASCII格式的inpcrd坐标文件读取坐标, 但不读取速度irest=0: 不重新启动模拟(不适用于最小化)maxcyc=2000: 最小化的最大循环数ncyc=1000: 最初的0到ncyc循环使用最速下降算法, 此后的ncyc到maxcyc循环切换到共轭梯度算法ntpr=100: 每ntpr次循环写入Ambermdout输出文件一次ntwx=0: 不输出Ambermdcrd轨迹文件(不适用于最小化)cut=8.0: 以埃为单位的非键截断距离(对于PME而言, 表示直接空间加和的截断. 不要使用低于8.0的值. 较高的数字略微提高精度, 但是大大增加计算成本)
升温输入
15. 创建包含以下升温设置的文件02_Heat.in:
| 02_Heat.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 | Heat
&cntrl
imin=0,
ntx=1,
irest=0,
nstlim=10000,
dt=0.002,
ntf=2,
ntc=2,
tempi=0.0,
temp0=300.0,
ntpr=100,
ntwx=100,
cut=8.0,
ntb=1,
ntp=0,
ntt=3,
gamma_ln=2.0,
nmropt=1,
ig=-1,
/
&wt type='TEMP0', istep1=0, istep2=9000, value1=0.0, value2=300.0 /
&wt type='TEMP0', istep1=9001, istep2=10000, value1=300.0, value2=300.0 /
&wt type='END' /
|
这些设置总结如下:
imin=0: 选择运行分子动力学(MD)[无最小化]nstlim=10000: 要运行的MD步数(运行时间长度为nstlim*dt, 单位ps)dt=0.002: 以皮秒(ps)为单位的时间步长. 每一MD步骤的时间长度ntf=2: 不计算受SHAKE约束的键所受的力ntc=2: 启用SHAKE来约束所有包含氢的键tempi=0.0: 初始恒温器的温度, 单位K(见NMROPT部分)temp0=300.0: 最终恒温器的温度 单位K(见NMROPT部分)ntwx=1000: 每ntwx步输出Amber轨迹文件mdcrd一次ntb=1: 等容的周期性边界ntp=0: 无压力控制ntt=3: 使用Langevin恒温器控制温度gamma_ln=2.0: Langevin恒温器的碰撞频率nmropt=1: 读入NMR限制和权重变化(见NMROPT部分)ig=-1: 随机化伪随机数发生器的种子(总是一个好主意, 除非你正在调试一个模拟问题)
通过NMROPT控制的恒温器温度
上面输入文件的最后三行允许恒温器在整个模拟过程中改变其目标温度. 对于前9000步, 温度将从0 K增加到300 K. 对于9001至10000步, 温度将保持在300 K.
成品模拟输入
警告: 就本身而言, 这个输入文件不适用于一般的MD模拟. 其中的NTPR和NTWX设置得非常低, 这样才可以对这个很短的模拟进行分析. 使用这样的设置进行更长时间的MD模拟会产生非常大的输出文件和轨迹文件, 并且比常规MD设置更慢. 对于真正的成品MD, 你需要增加NTPR和NTWX的值.
这个成品模拟的时间只有60 ps. 理想情况下, 我们需要运行这个模拟更长时间, 但为了节省完成本教程的时间, 我们限制了成品模拟的时间.
16. 创建包含以下成品MD设置的文件03_Prod.in:
| 03_Prod.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | Production
&cntrl
imin=0,
ntx=5,
irest=1,
nstlim=30000,
dt=0.002,
ntf=2,
ntc=2,
temp0=300.0,
ntpr=100,
ntwx=100,
cut=8.0,
ntb=2,
ntp=1,
ntt=3,
gamma_ln=2.0,
ig=-1,
/
|
成品模拟的设置总结如下:
ntx=5: 从无格式的inpcrd坐标文件中读取坐标和速度irest=1: 重新启动以前的MD运行(这意味着预期inpcrd文件中存在速度, 并将使用它们来提供初始原子速度)temp0=300.0: 恒温器温度. 在300 K运行ntb=2: 在恒定压力下使用周期性边界条件ntp=1: 使用Berendsen恒压器进行恒压模拟
输入文件
sander输入文件在这里下载: 01_Min.in, 02_Heat.in, 03_Prod.in
运行Amber MD模拟程序sander
现在我们有了所有的材料: 参数和拓扑文件prmtop, 坐标文件inpcrd和输入文件01_Min.in, 02_Heat.in, 03_Prod.in, 我们准备运行实际的最小化, 升温和成品MD.
为此, 我们将使用程序sander, Amber的通用MD引擎(也有一个高性能的版本, 称为pmemd, 它是商业版AMBER的一部分, 是MD引擎的最佳选择, 但只是用于教程的话, sander就足够了). sander从命令行运行. 在命令行上, 我们可以指定更多的选项, 并选择使用哪个文件用于输入.
17. 首先从终端将目录切换到Tutorial目录, 其中存放了所有的输入文件. ~是主目录的快捷方式, 其中有你创建的Tutorial目录.
cd ~/Tutorial
运行最小化
18. 用sander运行丙氨酸二肽的最小化
$AMBERHOME/bin/sander -O -i 01_Min.in -o 01_Min.out -p prmtop -c inpcrd -r 01_Min.rst -inf 01_Min.mdinfo
sander对MD模拟的每一步都使用一致的语法. 以下是sander命令行选项的总结:
-O: 覆盖输出文件, 如果它们已经存在-i 01_Min.in: 选择输入文件(默认mdin)-o 01_Min.out: 输出文件(默认mdout)-p prmtop: 选择参数和拓扑文件prmtop-c inpcrd: 选择坐标文件inpcrd-r 01_Min.rst: 输出包含坐标和速度的重启文件(默认restrt)-inf 01_Min.mdinfo: 输出包含模拟状态的MD信息文件(默认mdinfo)
取决于您计算机的性能, sander应该能在适量的时间(~27秒)内完成最小化.
sander运行完成后, 应该有一个输出文件01_Min.out, 一个重启文件01_Min.rst和一个MD信息文件01_Min.mdinfo. 您将使用重启文件01_Min.rst来升温系统.
最小化输出文件
最小化输出文件在这里下载: 01_Min.out, 01_Min.rst
19. 使用gedit打开输出文件01_Min.out
在01_Min.out文件中, 您可以找到最小化的详细信息. 在整个最小化过程中, 您应该能够看到系统能量ENERGY逐步降低.
运行升温MD
现在, 使用从开始的最小化得到的重启文件升温系统.
20. 使用sander升温丙氨酸二肽.
$AMBERHOME/bin/sander -O -i 02_Heat.in -o 02_Heat.out -p prmtop -c 01_Min.rst -r 02_Heat.rst -x 02_Heat.mdcrd -inf 02_Heat.mdinfo
以下是sander命令行选项的总结:
-c 01_Min.rst: 现在我们从最小化的重启文件获取输入坐标-x 02_Heat.mdcrd: MD模拟的输出轨迹文件(默认mdcrd)
取决于您的计算机性能, sander应该能在适量的时间内(约2.5分钟)完成升温模拟.
加热输出文件
加热输出文件可在这里下载. 有些文件已经压缩, 需要解压使用.
02_Heat.out, 02_Heat.rst, 02_Heat.mdcrd
21. 打开输出文件02_Heat.out查看系统输出.
在02_Heat.out文件中, 您能找到升温MD的输出. 您应该能够看到系统的信息, 包括每步的能量和温度. 例如在1000步的时候:
NSTEP = 1000 TIME(PS) = 2.000 TEMP(K) = 29.48 PRESS = 0.0
Etot = -6944.9552 EKtot = 112.3015 EPtot = -7057.2567
BOND = 1.0442 ANGLE = 1.7653 DIHED = 9.4906
1-4 NB = 2.6284 1-4 EEL = 46.3073 VDWAALS = 1448.7074
EELEC = -8567.1999 EHBOND = 0.0000 RESTRAINT = 0.0000
Ewald error estimate: 0.4641E-03
------------------------------------------------------------------------------
NMR restraints: Bond = 0.000 Angle = 0.000 Torsion = 0.000
===============================================================================
一些重要的数值包括:
NSTEP: MD模拟的时间步TIME: 模拟的总时间(包括重新启动)TEMP: 系统温度PRESS: 系统压力Etot: 系统的总能量EKtot: 系统的总动能EPtot: 系统的总势能
请注意, 因为升温中没有使用恒压器(压力控制), 所以压力为0.0.
运行成品MD
现在完成了最小化和升温. 我们继续开始实际的成品MD.
22. 使用sander运行丙氨酸二肽的成品MD
$AMBERHOME/bin/sander -O -i 03_Prod.in -o 03_Prod.out -p prmtop -c 02_Heat.rst -r 03_Prod.rst -x 03_Prod.mdcrd -inf 03_Prod.info &
注意: 命令的末尾加上了&, 这样sander将在后台运行
现在sander正在后台运行. 运行成品MD模拟需要一些时间.
但是我们想监控成品MD的状态. 所以我们将监控sander输出文件来检查运行状态. Linux程序tail可以输出文件的结尾部分.
23. 要监控作业的状态, 请使用程序tail将输出文件显示到终端
tail -f 03_Prod.out
当sander正在运行时, 上面的命令可以显示输出文件, 这对跟踪工作很有帮助. 您还可以监控mdinfo文件(cat 03_Prod.info), 该文件提供了详细的性能数据以及预计的完成时间.
24. 要退出tail, 按<CTRL-C>退出程序.
完成MD模拟
让MD模拟继续运行. 完成模拟需要一段时间(大约10分钟).
成品模拟输出
成品MD输出可以这里下载: 03_Prod.out, 03_Prod.rst, 03_Prod.mdcrd
一旦完成, 打开输出文件检查模拟是否正常完成.
25. 用gedit打开输出文件03_Prod.out, 查看MD模拟的输出.
可视化结果
你现在已经运行了一个MD模拟. 为了可视化结果, 我们现在将使用一个名为VMD(Visual Molecular Dynamics)的程序. 这是一个可以渲染3D分子结构的分子图形程序. VMD不仅可以加载蛋白质数据库(PDB)结构文件, 还加载许多程序的MD轨迹. (有关VMD的更深入教程可在教程主页找到).
26. 要启动VMD, 请打开一个终端, 将目录更改为Tutorial目录, 然后运行vmd.
记住~/Tutorial是您的Tutorial目录的快捷方式.
cd ~/Tutorial
vmd
VMD应该如下所示:
VMD是非常有用的工具, 用于可视化蛋白质, 核酸和其他生物分子的原子结构. 最常用的格式之一是PDB生物分子结构格式. 要加载一个PDB文件, 进入File -> New Molecule..., 然后选择PDB文件, 将其加载为一个New Molecule. VMD应该能够自动确定文件类型.
但是, 我们想要可视化丙氨酸二肽的轨迹. 现在我们将加载得到的MD轨迹来查看丙氨酸二肽的动态变化.
27. 使用File -> New Molecule...创建一个新的分子
28. 为New Molecule加载文件. 然后选择Amber参数和拓扑文件prmtop. 将文件类型设置为AMBER7.Parm. 点击Load.
29. 为0: prmtop加载文件. 然后选择Amber轨迹文件03_Prod.mdcrd. 将文件类型设置为AMBER Coordinates with Periodic Box. 点击Load.
VMD现在加载了您的要可视化的轨迹文件. VMD主窗口可用于控制播放.
在VMD的显示窗口中您应该能够看到丙氨酸二肽分子以及许多水分子. 您可以用鼠标旋转, 缩放和平移显示窗口中的分子.

许多不同的可视化选项可以在Graphics -> Representations窗口中进行更改.
可视化时也可以只显示丙氨酸二肽.
30. 在Selected Atoms中输入all not water
您可以将分子的绘图方法更改为更有趣的模型.
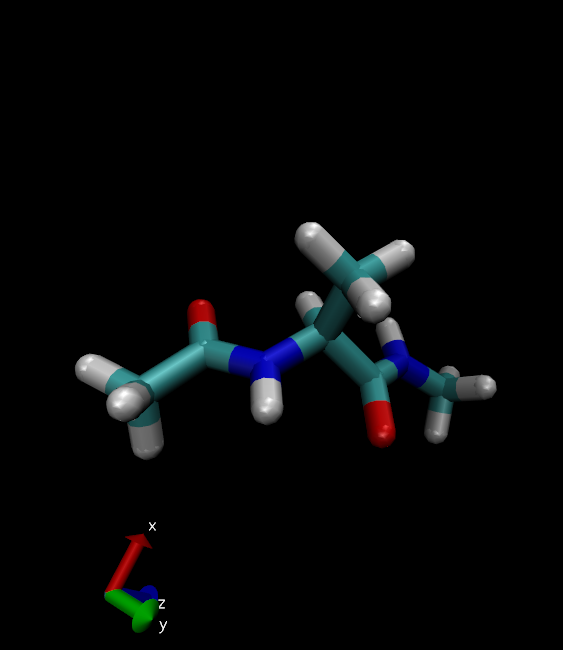
31. 在Drawing Method中选择Licorice
丙氨酸二肽看起来像这样:
更多VMD信息
VMD具有许多可用于分析和研究MD轨迹的功能. 例如, 可以对齐分子, 测量均方根偏差(RMSD), 保存轨迹中的结构, 测量整个轨迹中物理系统的参数. VMD也可以渲染一条轨迹的动画.
但是, 这些功能超出了本教程的范围. 更多详细信息请参阅AMBER教程主页面上的VMD教程.
分析MD结果
Amber包括一套工具用以检查和分析MD轨迹. 在本教程中, 我们将使用几个Amber程序做一些简单的分析并绘制结果. 分析主要是在终端的命令行中完成的.
32. 打开一个终端并将目录更改为存放教程文件的目录.
cd ~/Tutorial
33. 创建一个Analysis目录并切换到该目录.
mkdir Analysis
cd Analysis
现在我们将使用一个分析脚本process_mdout.perl来分析MD输出文件. 此脚本将从MD输出文件中提取能量, 温度, 压力, 密度和体积, 并将其保存到单独的数据文件中.
34. 使用process_mdout.perl处理MD输出文件
$AMBERHOME/bin/process_mdout.perl ../02_Heat.out ../03_Prod.out
现在很容易将保存在输出文件中的数据绘制出来.
我们将使用一个方便简单的绘图程序xmgrace自动绘制整个模拟过程中下面这些MD模拟性质的变化. 使用这个程序对我们自己比较方便, 你可以使用任何自己使用的绘图程序.
- MD模拟的温度
- MD模拟的密度
- MD模拟的总能量, 势能和动能.
但是, 对于MD模拟密度, 模拟的升温部分不包含密度输出. 您将需要编辑summary.DENSITY文件删除空白数据点, 这样xmgrace才能正常显示.
35. 使用gedit编辑summary.DENSITY文件以删除空白数据点(到20 ps).
36. 使用以下命令绘制模拟过程中性质的变化:
xmgrace summary.TEMP
xmgrace summary.DENSITY
xmgrace summary.ETOT summary.EPTOT summary.EKTOT
分析数据文件
分析数据文件可在这里下载: summary.TEMP, summary.DENSITY, summary.ETOT, summary.EPTOT, summary.EKTOT
成品MD系统的性质
警告: 我们应该运行这个模拟更长时间, 这样密度才能达到平衡, 并且模拟收敛. 然而, 为了节省完成本教程的时间, 成品MD模拟的时间设置得非常短, 这样才可以尽快分析结果.
得到的图应该看起来类似于下面这些:
丙氨酸二肽MD温度
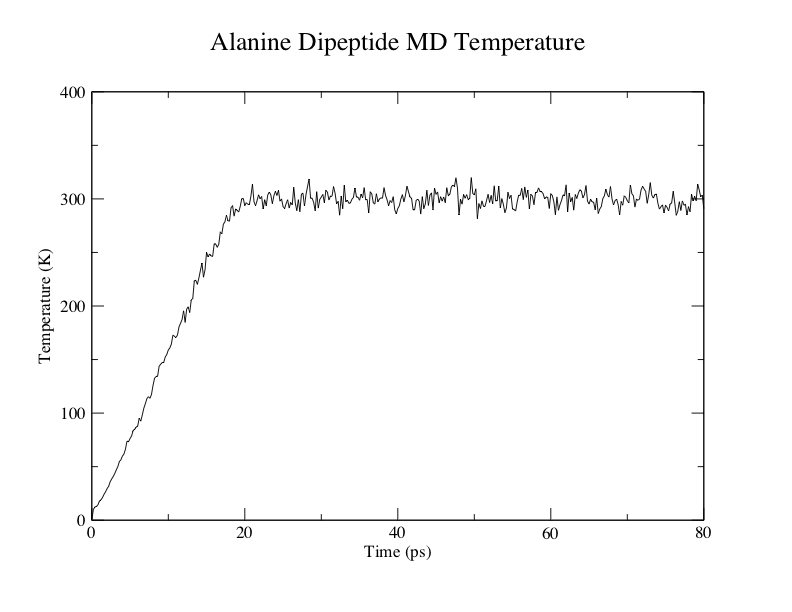
在这里你可以看到加热过程中温度线性增加(0-20 ps). 随后的成品模拟过程中温度波动相对稳定, 约为300 K.
丙氨酸二肽MD密度
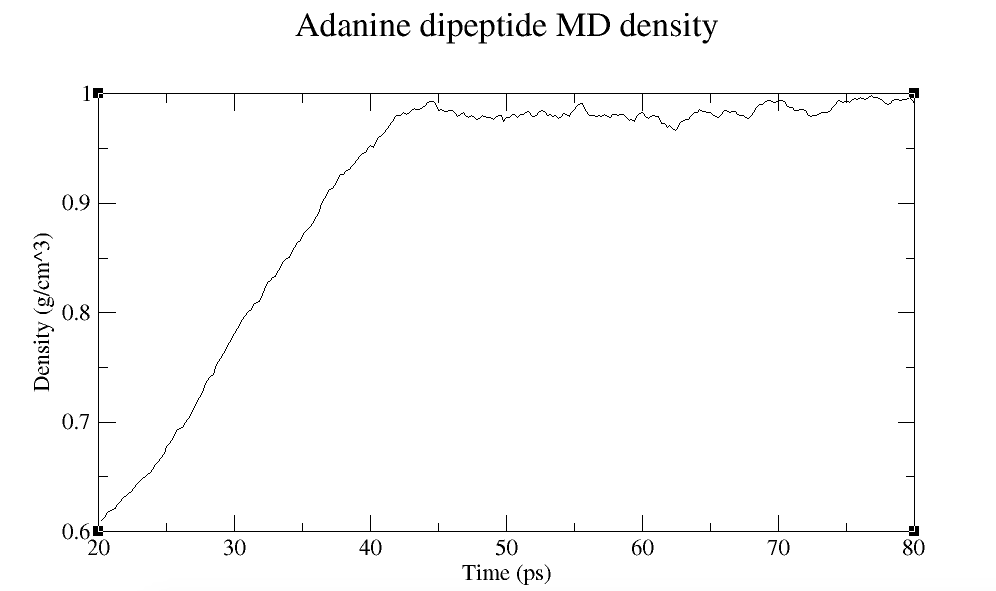
在20-80 ps时, 密度达到约1g/cm3. 当系统密度收敛时, 这对应于周期性盒子尺寸的变化.
丙氨酸二肽MD总能量, 势能和动能
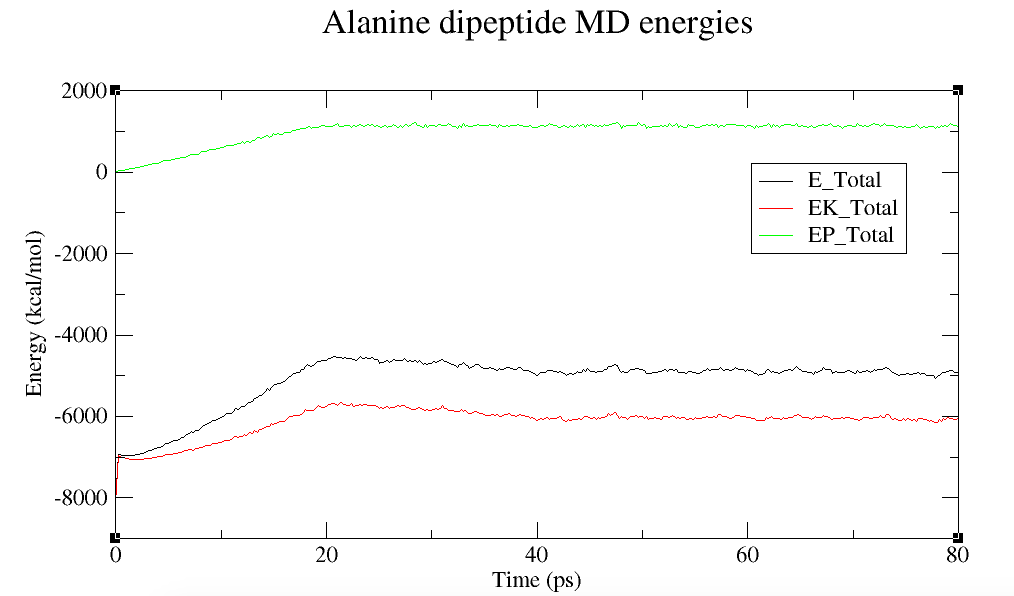
该图显示总的系统能量可以分解为总势能和总动能.
使用cpptraj分析RMSD
均方根偏差(RMSD)的值衡量了结构内部原子的坐标相对于某些参考分子坐标的相似程度. 对于这个例子, 我们将测量内部原子坐标相对于最小化结构的变化. 具体来说, 我们将分析丙氨酸的原子(残基2).
为了进行这个分析, 我们将使用cpptraj, 一个相当全面的处理MD轨迹的分析程序. 该程序可以运行用户编写的简单脚本, 其中指定要加载的轨迹, 要运行的分析以及要保存的处理过的轨迹或结构.
首先, 我们需要编写一个简单的cpptraj脚本来进行这个分析.
37. 使用gedit创建一个名为rmsd.cpptraj的cpptraj脚本.
trajin 02_Heat.mdcrd
trajin 03_Prod.mdcrd
reference 01_Min.rst
autoimage
rms reference mass out 02_03.rms time 2.0 :2
这是cpptraj脚本的简要总结:
trajin 02_Heat.mdcrd: 加载轨迹02_Heat.mdcrdreference 01_Min.rst: 定义结构01_Min.rst作为参考结构center: 1-3 mass origin: 将残基1-3的质心置于体系原点image origin center: 使用分子的质心将原子映像到原点rms reference mass out 02_03.rms time 2.0 :2: 使用参考结构计算质量加权的RMSD并输出到02_03.rms
cpptraj输入脚本文件
cpptraj输入脚本文件在这里下载: rmsd.cpptraj
运行cpptraj
要实际运行cpptraj, 我们必须从prmtop, mdcrd和参考rst文件所在目录中使用终端运行它.
38. 使用终端, 将目录切换到您的Tutorial文件夹, 然后运行cpptraj.
必须指定prmtop文件和cpptraj脚本.
cd ~/Tutorial
$AMBERHOME/bin/cpptraj -p prmtop -i rmsd.cpptraj &> cpptraj.log
现在我们的RMSD数据存储在文件02_03.rms中. 我们可以简单地使用xmgrace绘制这个文件.
39. 使用xmgrace绘制RMSD.
xmgrace 02_03.rms
Cpptraj输出文件
cpptraj输出文件在这里下载: 02_03.rms, cpptraj.log
丙氨酸二肽相对于最小化初始结构的MD RMSD
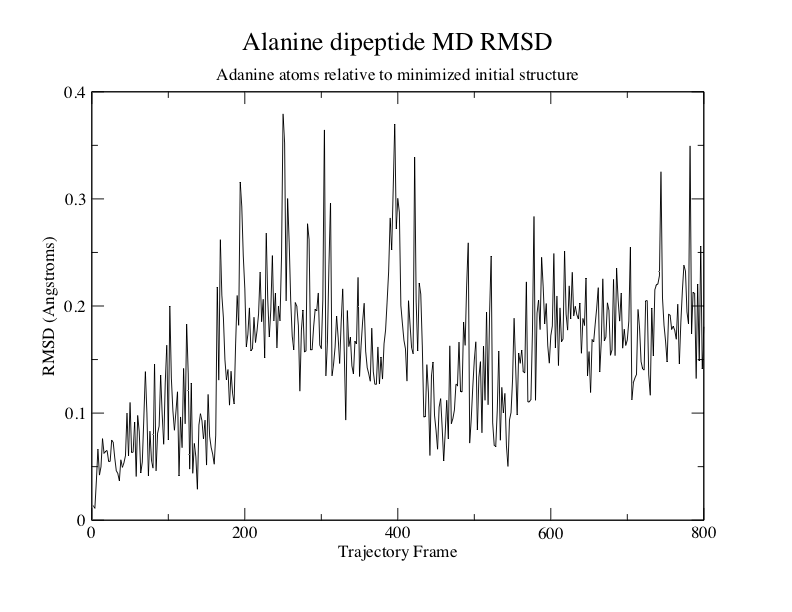
在这个例子中, 丙氨酸二肽的Phi/Psi二面角没有显著的构象变化. 这表明肽结构更稳定.
结论
恭喜! 您现在已经运行了您的第一个完整的MD模拟, 并成功地分析了结果. 这是设置, 运行和分析您自己的MD模拟的工作流程的一个相当简单的例子. 如果您想学习更多, 可以到AMBER网站上完成其他教程.
附录: 文件
本教程的用到的所有文件都可以在这里下载: Alanine_Dipeptide_Files.zip
