本教程一定程度上已经为另一教程替代, 但我们仍保留了它, 因为确实展示了一个有用的例子, 用于创建含有金属原子的体系.
你需要模拟的蛋白体系经常会含有非标准残基, 例如辅酶或抑制剂. 在这种情况下, 没有简单的办法构建拓扑和坐标文件. 你首先需要在xleap中构建一个新的组件, 添加上缺失的参数和电荷, 然后才能生成prmtop和inpcrd文件. 如果非标准残基是一个单独的分子, 你可以使用Antechamber(见基础教程B4 ). 然而, 在本教程中, 我们将要对质体蓝素蛋白(plastocyanin)进行建模, 其中含有一个铜原子, 结合到邻近的四个残基上. 本教程给出了如何在xleap中构建这样的残基的一个示例.
本教程有两个版本, 简单版本和高级版本. 在简单版本中仅仅创建一个新的铜残基, 假定为电荷+1的离子. 在高级版本中, 会创建特殊的HIS组胺酸残基和MET蛋氨酸残基, 这样就可以使用不同的电荷和键/角以及二面角参数.
AMBER高级教程A1: 模拟含有非标准残基的溶剂化蛋白(简单版本)
- 原始文档: Ross Walker, TUTORIAL A1: Simulating a Solvated Protein that Contains Non-Standard Residues(Simple Version)
- 参考: 原生態, Amber学习第五天: 模拟含有非标准残基的溶剂化蛋白
- 2018-01-31 06:31:20 翻译: 许楠(浙江大学); 校对: 康文渊(湖南大学)
介绍
前面我们研究的系统只含有标准的氨基酸或核苷酸残基, 所以我们不需要创建非标准的残基单元并为之提供自定义的参数. 本章我们将介绍一种创建非标准残基的方法. 需要注意的是, AMBER中有很多不同的方法可以实现这个目的, 这里的方法并不是唯一的. 另外, 请注意, 本教程提供的是一个含有铜离子的蛋白质的简单模拟示例. 这里做了很多假设, 例如铜的电荷仅仅用+1进行处理, 并且假设铜周围残基与铜结合后自身的参数没有发生变化. 严格来讲这并不正确, 但对于首次尝试是很有帮助的. 如果最终需要完成的是更加严谨的模拟, 那么就很有必要为与铜结合的残基创建新的单元, 并且为这些部分重新拟合电荷, 同时也要为那些受铜原子影响的键, 角和二面角提供新的参数.
在本教程中我们打算完成质体蓝素(Plastocyanin)蛋白在显式水溶剂中的模拟. 为此我们需要先做完很多事情:
- 在质体蓝素蛋白中有一个铜离子(Cu)与四个氨基酸相结合: His37, Cys84, His87和Met92. 为了将这些残基与铜离子结合在一起, 我们需要修饰这些残基并且为这些新产生的键类型(和与新键对应的角和二面角类型)提供参数.
- 质体蓝素的PDB文件(1PLC)中包含了结晶水, 我们应该保留. 但是PDB文件中只指定了氧的位置, 所以我们用
Xleap来添加丢失的质子. 我们会在运行成品模拟前使用能量最小化优化这些位置. - 有些异常的是该PDB文件含有一些显式的蛋白质上的质子. 这跟之前的情况也比较类似, 这些质子的命名约定与IPUAC的规定不符合. 既然这样, 我们就移除它们并让
Leap为我们自动添加质子就可以了. - 使用最可能的质子化态(处于中性pH)将会导致质体蓝素产生-9的净电荷, 因此我们就需要增加9个Na+补偿离子到体系中从而中和它的净电荷.
这有不少的工作量. 但是相对于使用AMBER 8之前的版本, 现在的工作已经简单多了. 本教程包含以下四个部分:
- 对PDB文件做一些修改
- 创建非标准
CUA单元 - 载入蛋白的PDB并创建库文件
- 生成
prmtop和inpcrd文件
第1步: 对PDB文件做一些修改
我们即将使用的PDB文件为PDB ID: 1PLC - 1PLC.pdb. 你应该读读PDB文件里的头部信息, 因为它经常包含了诸如无序对之类的信息. 通常一个PDB文件还可以包含结合与同一蛋白的一系列不同结构/抑制剂等. 在本PDB中, 我们可以看到#187和#183的水分子构成了无序对, 它们不可能同时存在.
| 1PLC.pdb | |
|---|---|
1 2 3 | REMARK 4 *1PLC 83*
REMARK 4 HOH 187 AND HOH 183 FORM A DISORDERED PAIR AND BOTH ARE NOT *1PLC 84*
REMARK 4 PRESENT SIMULTANEOUSLY. *1PLC 85*
|
然后我将随机地删除了#187水并保留#183. 我也将删除PDB尾部XLeap不会用到的成键信息. 这里我们的每个水分子都是一个独立的残基, 通常我们需要在残基之间添加TER标签, 从而确保它们不会连在一起形成长链. 幸运的是我们不需要为这里的结晶水添加标签, 因为Xleap足够聪明, 它知道我们的水是单独的分子, 这是水分子已经被定义为WAT单元的缘故.(注: 如果用不同的溶剂, 必须增加TER标签)
由于PDB文件不区分参与成键或发生了其他变化的半胱氨酸残基(因此硫原子上没有了氢), 因此我们需要编辑半胱氨酸残基才能获得正确的结果. 正常质子化的半胱氨酸残基在Leap中使用的名字为CYS, 去质子化的和/或与金属离子结合的残基为CYM, 而那些涉及到二硫键和其它键的残基为CYX. 由于质体蓝素蛋白中的84号半胱氨酸与铜离子结合, 因此我们需要将第84位残基的名字由CYS改为CYM. 对于组氨酸同样如此, 它可以在delta位置质子化(形成HID), epsilon位置质子化(HIE)或者在两处都质子化(HIP). 幸运的是在质体蓝素蛋白中处理组氨酸相当简单, 因为它就只有两个组氨酸残基(37和87)而且它们都通过delta氮与铜结合. 它们肯定都是在epsilon氮上发生质子化. 所以我们将两个组氨酸残基(37和87)的名字由HIS改为HIE. 接着Leap将会在正确的位置添加准确数量的质子. 这里是现在已经修改好了的PDB文件: 1PLC_mod.pdb
我们在使用该PDB文件之前, 还需要挑出它的非标准氢原子名称. 尽管可能通过质子化可以纠正这些非标准的氢原子名称, 即把它们从NMR中的命名约定转换到PDB中的约定, 但是经验显示NMR结构中的氢原子位置未必总是可靠的. 因此最佳的选择就是移除所有的质子, 然后允许Leap将它们在标准的位置添加回来. 因为我们通常在跑动力学之前会对系统进行能量最小化, 所以以上操作应该不会造成任何问题.
这里是已经移除了质子的PDB文件: 1PLC_mod2.pdb
下一步是弄懂我们将怎样处理铜原子. 在本例中我们将简单化处理, 仅仅把铜当作与周围残基结合的+1价离子处理. 理想情况下我们应该为四个与铜结合的残基创建新的单元, 然后为这些残基和铜原子重新拟合电荷和力场参数. 但是, 这是一项工作量很大的工作, 远远超出了本教程的范围. 为此我们应当尽可能简单地处理铜原子. 于是, 我们给铜原子自身构造了一个叫CUA的残基(注: 在选择残基名称时, 什么都行, 只要它是3个字符长, 并且没有在使用即可…你可以在Leap中使用list命令来检查当前都有哪些残基名字正在使用. ).
因此我们将编辑我们的PDB文件, 将铜残基的名称改为CUA.
| 1PLC_mod2.pdb | |
|---|---|
1 2 3 4 5 6 7 8 | ATOM 1538 OD1 ASN 99 7.523 13.716 33.177 1.00 33.45 1PLC1661
ATOM 1539 ND2 ASN 99 8.763 14.800 34.732 1.00 31.51 1PLC1662
ATOM 1540 OXT ASN 99 8.932 10.327 32.908 1.00 32.10 1PLC1663
TER 1547 ASN 99 1PLC1670
HETATM 1548 CU CUA 100 7.050 34.960 18.716 1.00 8.78 1PLC1671
HETATM 1549 O HOH 101 17.504 16.825 14.073 1.00 20.28 1PLC1672
HETATM 1550 O HOH 102 18.877 15.088 18.086 1.00 22.16 1PLC1673
HETATM 1551 O HOH 103 11.165 21.823 31.513 1.00 16.99 1PLC1674
|
原PDB文件中也包含了一些残基的几个”替代”结构.例如, 对于LYS30, 我们有:
| 1PLC_mod2.pdb | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 | ATOM 426 N LYS 30 -0.930 27.774 20.957 1.00 8.07 1PLC 549
ATOM 427 CA LYS 30 -2.028 28.602 20.421 1.00 10.86 1PLC 550
ATOM 428 C LYS 30 -1.629 30.029 20.254 1.00 9.16 1PLC 551
ATOM 429 O LYS 30 -1.226 30.665 21.243 1.00 7.63 1PLC 552
ATOM 430 CB ALYS 30 -3.201 28.489 21.415 0.50 13.41 1PLC 553
ATOM 431 CB BLYS 30 -3.250 28.517 21.354 0.50 15.09 1PLC 554
ATOM 432 CG ALYS 30 -4.397 29.366 21.249 0.50 16.84 1PLC 555
ATOM 433 CG BLYS 30 -4.600 28.495 20.646 0.50 21.50 1PLC 556
ATOM 434 CD ALYS 30 -5.681 28.891 21.893 0.50 20.64 1PLC 557
ATOM 435 CD BLYS 30 -5.745 28.171 21.589 0.50 24.43 1PLC 558
ATOM 436 CE ALYS 30 -5.527 28.212 23.225 0.50 23.18 1PLC 559
ATOM 437 CE BLYS 30 -5.585 26.973 22.460 0.50 24.88 1PLC 560
ATOM 438 NZ ALYS 30 -6.825 28.052 23.929 0.50 20.02 1PLC 561
ATOM 439 NZ BLYS 30 -5.971 25.681 21.860 0.50 26.52 1PLC 562
ATOM 440 H LYS 30 -0.661 27.945 21.802 1.00 8.96 1PLC 563
ATOM 441 HA LYS 30 -2.302 28.229 19.582 1.00 10.61 1PLC 564
ATOM 442 1HB ALYS 30 -3.380 27.572 21.665 0.50 14.09 1PLC 565
ATOM 443 1HB BLYS 30 -3.134 27.712 21.919 0.50 15.93 1PLC 566
ATOM 444 2HB ALYS 30 -2.700 28.880 22.297 0.50 13.96 1PLC 567
ATOM 445 2HB BLYS 30 -3.188 29.326 21.939 0.50 15.16 1PLC 568
ATOM 446 1HG ALYS 30 -4.210 30.316 21.598 0.50 18.44 1PLC 569
ATOM 447 1HG BLYS 30 -4.784 29.428 20.234 0.50 20.74 1PLC 570
ATOM 448 2HG ALYS 30 -4.609 29.538 20.265 0.50 17.98 1PLC 571
ATOM 449 2HG BLYS 30 -4.629 27.910 19.855 0.50 20.04 1PLC 572
ATOM 450 1HD ALYS 30 -6.278 29.712 22.058 0.50 21.47 1PLC 573
ATOM 451 1HD BLYS 30 -5.929 28.976 22.170 0.50 24.46 1PLC 574
ATOM 452 2HD ALYS 30 -6.224 28.346 21.263 0.50 21.94 1PLC 575
ATOM 453 2HD BLYS 30 -6.606 28.094 21.037 0.50 24.54 1PLC 576
ATOM 454 1HE ALYS 30 -5.138 27.298 23.130 0.50 22.69 1PLC 577
ATOM 455 1HE BLYS 30 -4.709 26.867 22.883 0.50 25.84 1PLC 578
ATOM 456 2HE ALYS 30 -4.956 28.735 23.845 0.50 23.28 1PLC 579
ATOM 457 2HE BLYS 30 -6.257 27.063 23.262 0.50 25.75 1PLC 580
ATOM 458 1HZ ALYS 30 -7.360 28.765 23.779 0.50 21.25 1PLC 581
ATOM 459 1HZ BLYS 30 -6.721 25.740 21.353 0.50 26.03 1PLC 582
ATOM 460 2HZ ALYS 30 -7.206 27.240 23.677 0.50 21.83 1PLC 583
ATOM 461 2HZ BLYS 30 -5.279 25.211 21.533 0.50 25.17 1PLC 584
ATOM 462 3HZ ALYS 30 -6.682 27.986 24.852 0.50 21.60 1PLC 585
ATOM 463 3HZ BLYS 30 -6.289 25.095 22.599 0.50 25.92 1PLC 586
|
Leap软件会默认使用A构型并忽略其它的. 这对我们的目的而言还可以. 特别地, 如果我们想要从其它构型之一开始, 我们就需要从文件中移除A构型.
最后我们需要做的就是在我们的铜原子和第一个结晶水之间加上TER标签. 由于铜原子严格来说并不是蛋白质链的一部分, 我们将在Leap中手动给铜原子添加键. 因此, 铜原子任何一侧的TER标签将会制止Leap程序试图将其作为蛋白质的一部分而造成混乱.
这里是经过以上修改后的PDB文件: 1PLC_mod_final.pdb
第2步: 创建非标准CUA单元
如果此刻我们简单地将编辑后的PDB文件加载到XLeap中, 那么该文件的绝大部分都会正常加载. 但是我们非标准CUA残基将会出现问题. 我们需要告诉XLeap我们的非标准单元是什么, 这样才能够顺利地将1PLC_modified_final.pdb加载到XLeap中.
我们有几个选择来处理这个问题. 对于一个简单的分子, 最简单的选择就是使用Antechamber, 就像在教程5中所做的那样. Antechamber提供了一个自动创建非标准的单元的方法. 然而, 它只适用于完整的分子, 而不是我们这里的分子片段.
第二个选择是使用Xleap简单地编辑一下这些残基, 然后我们退出时忽略它们. 对于铜原子这种简单的情况, 这是最快的方法, 但是没法重新使用这些残基. 例如, 如果我们想要创建第二个非常相似的蛋白质, 我们将不得不在Xleap中的重新来过, 重复地编辑这些残基. 虽然仅仅处理铜这并不成问题, 但如果我们掌握了一种可以移植到更复杂系统的处理方法, 那也许是最好的.
第三个选择, 也就是我们在这里将会用到的方法, 即为非标准单元创建一个新的库文件. 通过这种方式, 我们可以重新使用该单元. 对于相似的蛋白质, 我们只需在加载蛋白质PDB之前将库文件加载到Xleap中就可以了. 对于大的辅酶, 如NADH, 这是推荐使用的方法.
所以, 如果你还没有这么做, 那就启动Xleap并且把我们的 1PLC_mod_final.pdb文件加载到一个新的称为PLC的单元. (译注: 现在AMBER14版本之后蛋白质力场变成leaprc.protein.ff14SB这样的格式)
$AMBERHOME/exe/xleap -s -f $ AMBERHOME/dat/leap/cmd/leaprc.ff99
> 1PLC = loadpdb 1PLC_mod_final.pdb
我们应该检查Xleap给我们的信息, 确保它完成了我们所期望的. 它应该报告只有一个未知的残基, 也就是我们的CUA. 它也应该添加了所有缺失的质子. 因此, 我们预计Xleap会添加总计922个H原子. 请确认情况确实如此. 如果不是, 那么你在在PDB的编辑过程中遗漏了一些东西. 同时, 也会有一些关于重复的原子名称的警告, 这是因为我们有几个残基拥有”替代”结构. 这些只是警告, 我们不需要担心, Leap只会使用第一种结构. 这只是一个检查一切正常的测试. 我们现在可以退出Xleap了.
创建CUA单元
现在我们需要为CUA单元创建一个库文件. 因为我们需要这个单元的初始结构, 所以最简单的处理方法就是简单地把它从1PLC_modified_final.pdb文件中剪切出来, 并把它保存为它自己的PDB文件. 这样处理铜可能看起来很疯狂, 实际上可能就是这样, 但是通过这样做, 你学习到了一种可以移植到很复杂系统的方法.
现在我们可以继续往下做并再次加载Xleap(你是否已经熟悉它了?:-))并将PDB文件加载到它自己的单元中:
$AMBERHOME/exe/xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99
> CUA = loadpdb cua.pdb
现在我们需要告诉Xleap关于我们的非标准残基的信息. 如果我们有好几个原子, 我们需要告诉它哪个原子与哪个原子相连, 我们可以在Xleap的编辑窗口中手工完成, 或者在Leap中使用bondByDistance命令. 但是, 由于我们的铜单元只包含一个原子, 所以我们可以跳过这一步.
> edit CUA
这应该会在编辑窗口中显示我们的CUA残基, 您可以在“Dispaly”菜单中打开原子名称显示.
下一步是具体指定原子类型和电荷.
通常情况下, 你需要计算铜原子和与其相连的残基中其它所有原子的原子电荷. 这是很有必要的, 因为铜原子的存在将改变这些周围残基中的电子分布. 对于AMBER, 使用约束静电势法(RESP)就可以完成. 计算RESP电荷的细节可以在AMBER的网站上找到. 在本教程中我不会介绍这一步. 相反, 我们只是假定铜原子具有+1的电荷, 并且不影响相邻的单元. 请注意, 现在RESP的拟合程序已经实现了自动化, 并且有一个名为RED的免费程序, 想要详细了解如何获取和使用RED, 请访问 http://upjv.q4md-forcefieldtools.org/RED/.
对本教程而言, 我们将使用下面屏幕截图中显示的电荷. 为了指定电荷和原子类型, 我们需要选择整个单元. 点击操作栏(Manipulation bar)中的选择按钮, 然后框选整个分子. 它的颜色应该发生了变化. 然后进入编辑菜单: (译注, 键盘的NUM LOCK打开可能会导致菜单栏失效) Edit->Edit Selected Atoms
应该会出现下面的框.
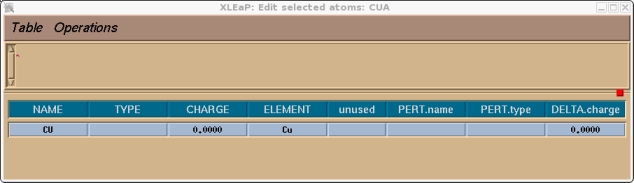
现在你应该检查一下并为该单元中的所有原子指定一个原子类型和电荷, 在本例中就只有铜原子. 我们将选择目前没有被使用过的CU作为铜的原子类型.
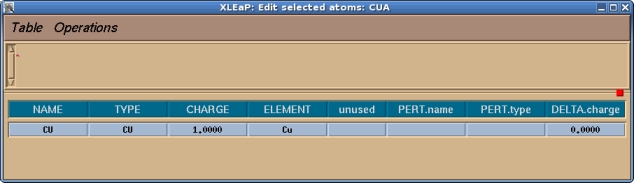
一旦完成, 我们就可以选择Table->Save and Quit. 然后我们可以关闭编辑窗口, 只需要留下命令窗口即可. 通常, 在我们可以为已修改好的单元生成库文件之前, 需要做的最后一件事情就是告诉Xleap这个单元的头部原子是什么以及尾部原子是什么, 这些信息用于连接蛋白质的骨架. 例如, 如果我们输入desc MET, 我们将看到MET有一个定义好的头部和尾部:
> desc MET
UNIT name:
Head atom: .R<MET 1>.A<N 1>
Tail atom: .R<MET 1>.A<C 16>
Contents:
R<MET 1>
但是, 如果我们查看CUA, 就会发现没有定义头和尾原子.
> desc CUA
UNIT name: CUA
Head atom: null
Tail atom: null
Contents:
R<CUA 100>
因为我们不会将铜残基作为蛋白质链的一部分, 所以这没有问题, 但是, 如果你要创建一个成为蛋白质链的一部分的残基, 你需要使用Leap中的set命令来指示哪个原子是新单元中的头部原子, 哪个是尾部原子.
我们现在可以保存已完成的库文件:
> saveoff CUA cua.lib
这里就有: cua.lib
第3步:载入蛋白的PDB并创建库文件
现在差不多快要完成了. 我们已经定义了非标准残基, 现在只需要让Xleap知道还缺少哪些参数. 在此之前, 让我们先在Xleap中为蛋白质添上必要的键, 这样我们就可以让Xleap告诉我们哪些参数缺失了.
$AMBERHOME/exe/xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99
为了让Xleap在加载1PLC_mod_final.pdb文件时认识我们的新残基, 我们需要确保首先加载了新的库文件. 这样将会定义CUA单元, 于是当Xleap在PDB文件中遇到它时, Xleap将会知道它的拓扑, 电荷和原子类型:
> loadoff cua.lib
> 1PLC = loadpdb 1PLC_mod_final.pdb
现在应该没有了错误信息. 只有关于原子名称重复的警告.
在我们进一步处理之前, 我们必须确保所有与铜原子相连的键都被定义好了. 目前它们还没有被定义. 我们需要在铜和半胱氨酸(84)的硫原子之间, 铜和MET(92)硫原子之间以及铜和两个组氨酸(37和87)的delata氮之间添加键. 我们可以用bond命令来完成, 或者可以编辑1PLC并手动画出键, 但是编辑一个蛋白质时, 这是非常乏味的, 尝试一下, 你会明白我的意思.
> bond 1PLC.37.ND1 1PLC.100.CU
> bond 1PLC.87.ND1 1PLC.100.CU
> bond 1PLC.84.SG 1PLC.100.CU
> bond 1PLC.92.SD 1PLC.100.CU
注意: desc命令可以列出残基编号和原子名称, 以便确定要成键的对象. 例如desc 1PLC.CUA.
这将会创建四个缺失的键. 我们现在可以继续往下做了, 为我们的系统创建一个截断八面体盒子并使用TIP3P水进行溶剂化. (译注: 需要先source leaprc.water.tip3p)
> solvateoct 1PLC TIP3PBOX 12
除了已经存在的结晶水外, 该命令将在我们的系统周围缓冲宽度(译注: 分子到盒子边缘的最小距离)为12埃的区域内添加其他的水分子. 接下来我们中和系统的电荷, 它目前有-9.0(check 1PLC)的净电荷. 因此, 我们应该添加总共9个Na+离子.
> addions 1PLC Na+ 9
这可能需要运行几秒钟的时间. 输出结果应该看起来像下面这样.
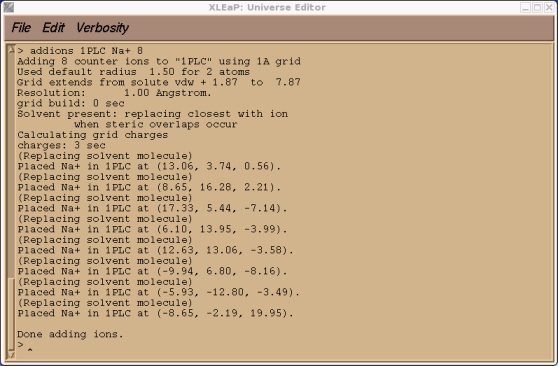
现在我们可以尝试保存我们的prmtop和inpcrd文件. 此时, 我们应会发现Xleap找不到类型CU.
> saveamberparm 1PLC 1PLC.prmtop 1PLC.inpcrd
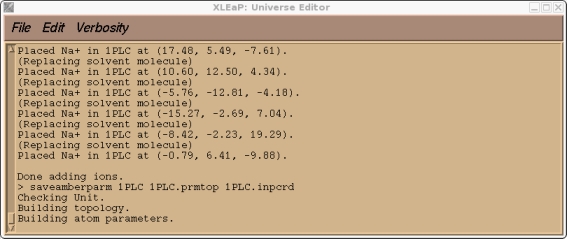
这是预料到的, 因为CU是一个我创建的原子类型, 目前在标准的PARM99力场中不存在. 如果你检查我们的1PLC单元, 你还应该会发现由于溶剂化而产生的大量”紧密接触”(译注: 原子间距过小), 以及大量与新的CU原子类型有关的参数缺失.
> check 1PLC
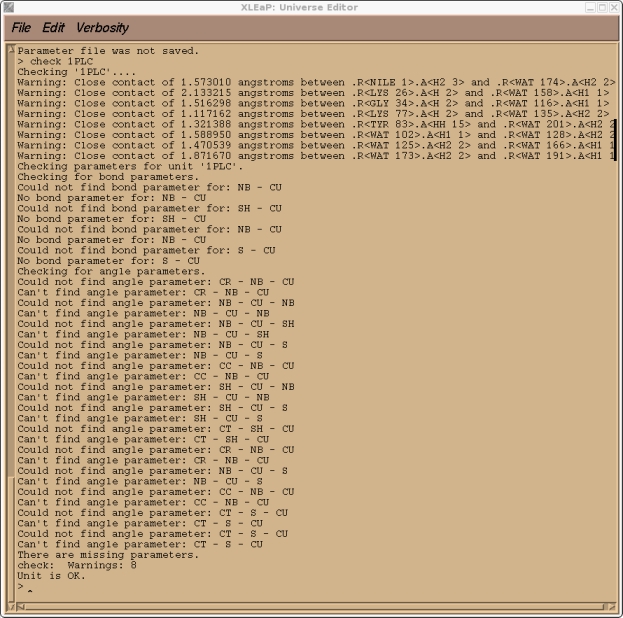
因此, 为了能继续向前推进, 我们需要将所有的这些参数添加到AMBER力场中. 然而, 在我们退出Xleap之前, 我们将保存一个1PLC库文件, 从而以后不必重复以上所做的所有步骤, 我们可以重新加载这个库文件.
> saveoff 1PLC 1PLC.lib
这里就有: 1PLC.lib
第4步: 生成prmtop和inpcrd文件
为了引入新的参数, 我们有两个选择. 我们可以修改核心力场文件或者可以创建一个frcmod文件, 其中包含针对此项目所做的更改. 第二个选择是一个更好的方案, 因为修改主文件可能会导致与使用相同安装软件的其他人发生冲突. 创建一套参数是一门艺术活, 因为人们往往碰到未知的参数(如本例中铜对其配体的力常数). 本教程的目的只是简单地介绍运行AMBER的机制, 所以接下来我会提供给你们所有的参数. 请注意, 这些仅供教程之用, 我并不保证这些参数的有效性和适用性. 文献中有大量涉及参数评价的文章, 用户们遇到不寻常的化学环境时也建议咨询他们. AMBER8手册中题为“参数开发”的12.1节可以作为一个起点.
这里就是我为质体蓝素创建的frcmod文件.
| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 | # modifications to force field for poplar plastocyanin
MASS
CU 65.36
BOND
NB-CU 70.000 2.05000 #kludge by JRS
CU-S 70.000 2.10000 #kludge by JRS
CU-SH 70.000 2.90000 #for pcy
CT-SH 222.000 1.81000 #met(aa)
ANGLE
CU-NB-CV 50.000 126.700 #JRS estimate
CU-NB-CR 50.000 126.700 #JRS estimate
CU-NB-CP 50.000 126.700 #JRS estimate
CU-NB-CC 50.000 126.700 #JRS estimate
CU-SH-CT 50.000 120.000 #JRS estimate
CU-S -CT 50.000 120.000 #JRS estimate
CU-S -C2 50.000 120.000 #JRS estimate
CU-S -C3 50.000 120.000 #JRS estimate
NB-CU-NB 10.000 110.000 #dac estimate
NB-CU-SH 10.000 110.000 #dac estimate
NB-CU-S 10.000 110.000 #dac estimate
SH-CU-S 10.000 110.000 #dac estimate
CU-SH-CT 50.000 120.000 #JRS estimate
CT-CT-SH 50.000 114.700 #met(aa)
HC-CT-SH 35.000 109.500
H1-CT-SH 35.000 109.500
CT-SH-CT 62.000 98.900 #MET(OL)
DIHE
X -NB-CU-X 1 0.000 180.000 3.000
X -CU-SH-X 1 0.000 180.000 3.000
X -CU-S -X 1 0.000 180.000 3.000
X -CT-SH-X 3 1.000 0.000 3.000
NONBON
CU 2.20 0.200
|
#之后的都为注释内容. 你可以看到我们指定了质量, 缺失的键, 角, 二面角及vDW参数.
现在我们可以将其加载到Xleap中, 它会把所有这些参数都添加到我们选用的PARM99力场中.
$AMBERHOME/exe/xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99
> loadamberparams plc.frcmod
> loadoff 1PLC.lib
这样我们就应该能够创建拓扑和坐标文件了.
> saveamberparm 1PLC 1PLC.prmtop 1PLC.inpcrd
这里就有这些文件: 1PLC.prmtop 1PLC.inpcrd
如果你想在VMD中观察这个初始结构, 我们可以使用ambpdb创建一个PDB文件, (译注, 如果想从重启文件生成PDB文件, 需要使用-c参数代替<)
$AMBERHOME/exe/ambpdb -p 1PLC.prmtop < 1PLC.inpcrd > 1PLC.inpcrd.pdb
这里就有一份: 1PLC.inpcrd.pdb
现在我们就可以使用这些文件进行质体蓝素的模拟了. 如果你愿意, 可以自己试试看. 记住, 该模拟是在显示溶剂和周期性盒子中进行, 所以你需要使用周期性边界条件. 起初, 你也需要将系统进行能量最小化以移除过小的原子间距. 接着我会将跑一个20 ps的升温过程, 在恒容周期性边界条件下从0 K升温到300 K, 然后转到300 K, 恒压条件下进行长时间的平衡模拟.
