- Daniel R. Roedaniel.r.roe@gmail.com, July 2014 原始文档
- 2018-03-15 21:47:26 翻译: 席昆(武汉大学)
注意: 本教程使用的是AmberTools14的CPPTRAJ
1. 简介
本教程描述的主要是MD模拟后最常用分析中的一种: 即坐标均方根偏差(RMSD). 此外还会标明在CPPTRAJ分析中读入的拓扑及参照文件.
RMSD是测量某部分特定原子相对于一参考结构的坐标偏差, 最完美的重合时则RMSD为0.0. RMSD定义为:
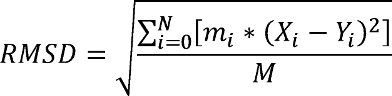
这里 $N$ 是分析原子的总数, $m_i$ 是第 $i$ 个原子质量, $X_i$ 与 $Y_i$ 分别表示相同坐标系下目标原子 $i$ 的坐标矢量及参考结构中原子 $i$ 的坐标矢量, $M$ 是总质量. 如果RMSD计算中不考虑质量加权, 则所有的 $m_i=1, M=N$.
当要计算相对于某参考结构的RMSD时, 需要满足以下重要的两点要求:
- 目标结构的原子数与参考结构原子数相同;
- 目标结构中原子顺序与参考结构原子顺序相同;
计算环境需求
本教程假定读者已安装且测试了AmberTools, 同时已完成CPPTRAJ教程C0. 另外, 需要xmgrace读取部分输出数据, 推荐使用分子可视化软件(如VMD或Chimera)来观察结构或运动轨迹.
本教程将分析beta-harpin trpzip的一段较短的模拟轨迹, 其格式为NetCDF. 该格式相比ASCII格式, 能使数据处理速度更快, 压缩后数据量更小, 数据有更高的精度且数据保存时更加稳定. NetCDF在Amber中默认支持, 如果你的轨迹无法打开, 请联系Amber mailing list寻求帮助. 分析所用的轨迹文件, 相关的拓扑文件以及其他文件可从以下链接下载:
trpzip2.gb.ncMD5 sum: 059435cfe1fcd57c327d46711da9cefftrpzip2.ff10.mbondi.parm7MD5 sum: 140044a45c683612da3431dfd0697e22trpzip2.1LE1.1.rst7MD5 sum: 9d4a64e65c77e9833ff38a424b4669d0trpzip2.1LE1.10.rst7MD5 sum: 6ea49fa08bae5fd92fb4246ee7ec34ca2GB1.pdbMD5 sum: 69fd269e026f25aef2a5e94654296ae5
关于CPPTRAJ的更多信息, 请查看:
2. 计算RMSD
在终端中输入cpptraj启动CPPTRAJ:
[user@computer ~]$ cpptraj
CPPTRAJ: Trajectory Analysis. V14.05
___ ___ ___ ___
| \/ | \/ | \/ |
_|_/\_|_/\_|_/\_|_
>
再读入拓扑及轨迹文件:
> parm trpzip2.ff10.mbondi.parm7
Reading 'trpzip2.ff10.mbondi.parm7' as Amber Topology
> trajin trpzip2.gb.nc
Reading 'trpzip2.gb.nc' as Amber NetCDF
指定RMSD的计算对象:
> rms ToFirst :1-13&!@H= first out rmsd1.agr mass
RMSD: (:1-13&!@H*), reference is first frame (:1-13&!@H*), with fitting, mass-weighted.
如上述指令, 我们计算了残基1-13中所有非氢原子的质量加权后的RMSD, 保存在名为ToFirst的数据集中, 参考结构是轨迹的第一个构象, 计算结果写入rmsdl.agr文件(这里文件后缀为.agr, 因此输出数据的格式为xmgrace). 注意, rms意指轨迹的所有结构将默认经过转动和平动与参考结构(第一个结构)达到最优重合后再计算RMSD. 因此, 除非命令中指定nofit, 否则rms指令对其他选项计算时会自动优化结构(即此计算).
命令行输入run开始轨迹处理, 处理完之后, 如果安装了xmgrace, 可直接在CPPTRAJ命令行查看结果:
> xmgrace rmsd1.agr
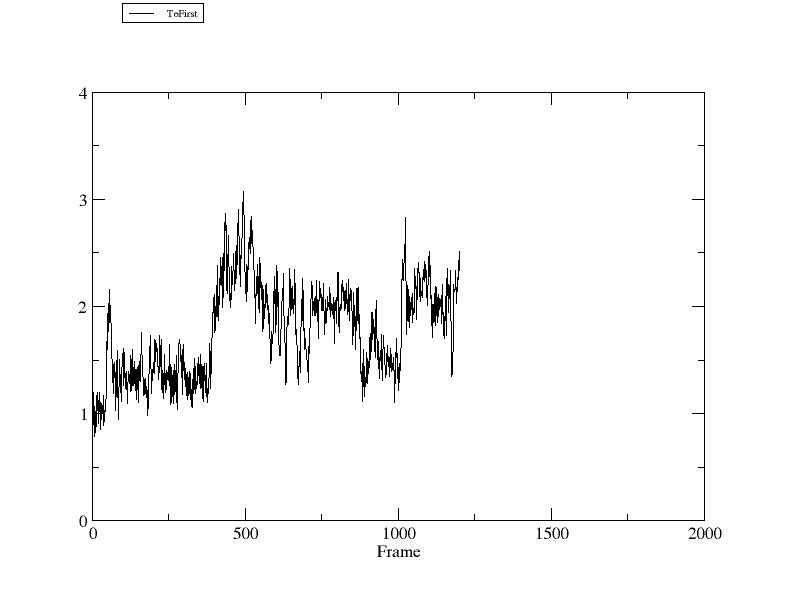
上图中横坐标是构象数, 纵坐标是相对第一个构象的RMSD(单位埃).
3. 导入选定的参考结构进行计算
相对于轨迹的第一个构象所计算的RMSD能说明与初始结构的偏差大小, 但如果要与某个如由实验得到的结构(例如: X射线或NMR测得的结构)进行比较, 则需要选定来确定参考结构. 本教程中由NMR得到的trpzip2结构PDB代码为1LE1, 而本教程中Amber重新分析时可以读入的文件trpzip2.1LE1.1.rst7包含了NMR实验所测的许多结构中的第一个结构, 这个结构通过reference指令就可以选作参考结构.
因为这个文件的拓扑与轨迹文件的拓扑相同, 此处无需再读入另一个新的拓扑文件.
> reference trpzip2.1LE1.1.rst7
Reading 'trpzip2.1LE1.1.rst7' as Amber Restart
'trpzip2.1LE1.1.rst7' is an AMBER restart file, no velocities, Parm trpzip2.ff10.mbondi.parm7 (reading 1 of 1)
类似的, Amber重新分析时可以读入的文件trpzip2.1LE1.10.rst7则是包含trizip2的NMR实验所测的许多结构中的第十个结构:
> reference trpzip2.1LE1.10.rst7 [tenth_member]
Reading 'trpzip2.1LE1.10.rst7' as Amber Restart
'trpzip2.1LE1.10.rst7' is an AMBER restart file, no velocities, Parm trpzip2.ff10.mbondi.parm7 (reading 1 of 1)
上述指令中, 参考结构用[tenth_member]标记. 在CPPTRAJ中, 可以用格式[<name>]来标记参考结构和拓扑文件. 之后即可通过读入文件的名称、序号(从0开始编号的读入文件)或特定的标记名来调用.
其中, CPPTRAJ中可以通过选择轨迹中的任一结构的构象编号或直接通过lastframe调用最后的结构选作参考结构. 例如, 我们可以使用关键词lastframe调用最后一个结构作为参考结构, 并标记为[last]:
> reference trpzip2.gb.nc lastframe [last]
Reading 'trpzip2.gb.nc' as Amber NetCDF
'trpzip2.gb.nc' is a NetCDF AMBER trajectory, Parm trpzip2.ff10.mbondi.parm7 (reading 1 of 1201)
使用list指令可以显示当前所有读入的参考结构的信息:
> list ref
REFERENCE FRAMES (3 total):
0: 'trpzip2.1LE1.1.rst7', frame 1
1: [tenth_member] 'trpzip2.1LE1.10.rst7', frame 1
2: [last] 'trpzip2.gb.nc', frame 1201
Active reference frame for masks is 0
如上显示为读入的参考结构的序号、文件名和特定的标记名(只显示确定被标记的).
最后, 在显示的所有参考结构下, Active reference frame for masks is 0表示distance-based masks将以序号0的参考结构来决定选定的原子. 在CPPTRAJ中, 使用当前结构来更新基于距离的masks的操作是mask action.
4. 计算相对于选定参考结构的RMSD
我们现在已经导入了一些参考结构, 接下来就可以分别计算相对于这些参考结构的RMSD, 有以下三种方式选择参考结构:
reference: 选择第一个读入的参考结构;refindex <#>: 使用参考结构的顺序编号<#>选择读入的参考结构(所以如果选择reference可以用refindex 0);ref <name | tag>: 使用参考结构的文件名或者标记名进行选择;
下一步参考上述三种参考结构的选择方式计算每一个参考结构对应的RMSD:
> rms ToMember1 :1-13&!@H= reference out rmsd2.agr
Mask [:1-13&!@H*] corresponds to 116 atoms.
RMSD: (:1-13&!@H*), reference is reference frame trpzip2.1LE1.1.rst7 (:1-13&!@H*), with fitting.
> rms ToMember10 :1-13&!@H= refindex 1 out rmsd2.agr
Mask [:1-13&!@H*] corresponds to 116 atoms.
RMSD: (:1-13&!@H*), reference is reference frame trpzip2.1LE1.10.rst7 (:1-13&!@H*), with fitting.
> rms ToLast :1-13&!@H= ref [last] out rmsd2.agr
Mask [:1-13&!@H*] corresponds to 116 atoms.
RMSD: (:1-13&!@H*), reference is reference frame trpzip2.gb.nc (:1-13&!@H*), with fitting.
注意每一个参考结构的相应RMSD计算中, rms计算得到的结果都保存在相同命令的文件中, 在CPPTRAJ所有的指令都会有out选项控制输出;
输入run完成轨迹处理后, 如果安装了xmgrace则可以直接在CPPTRAJ中输入下面指令参看结果:
> xmgrace rmsd2.agr
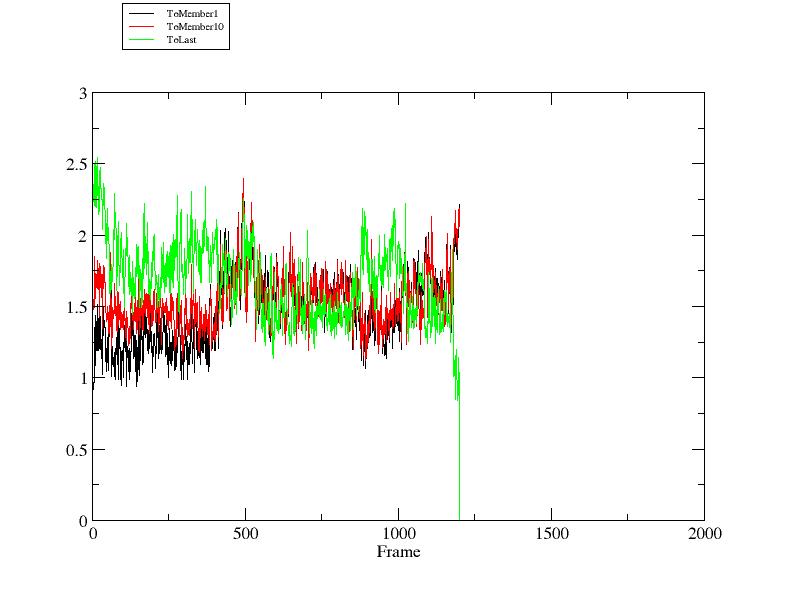
我们发现轨迹结构在初始阶段相比NMR实验得到的第十个结构(member 10), 其与第一个结构(member 1)更加相近.
5. 不同拓扑下的RMSD计算
计算有不同拓扑的结构之间RMSD在分析中常常很有用. 例如: 与本教程中的分子的链有相似疏水残基排列的一个相关发夹(harpin)结构(GB1, PDB ID: 2GB1), 其来自于G蛋白的B1 IgG-结合域. 但不同的是此相关结构在弯曲区域有另外两个残基. 有人可能就会想了解trpzip2与GB1链的结构相似程度或者想对其对应结构进行比对, 这就需要读入GB1作为参考结构, 同时将trpzip2与GB1相同的结构部分作单独的特定标记. 因此检查这两个分子结构后, 发现trpzip2的残基1-6和7-12与GB1的残基42-47和50-55基本相叠合.
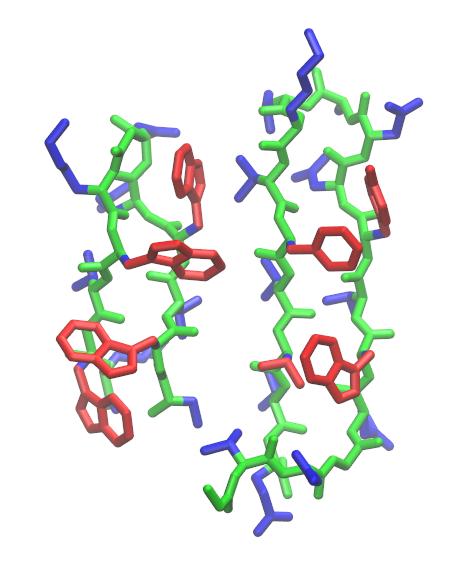
Trpzip2 (左侧) 与GB1第二个发夹(右侧, PDB代码: 2GB1). trpzip2的疏水残基架构与GB1的相对应, 如红色显示, 上图用VMD 1.9.1产生.
由于GB1与trpzip2分子的拓扑不同, 因此需要为GB1单独读入新的拓扑文件.
> parm 2GB1.pdb
Reading '2GB1.pdb' as PDB File
2GB1.pdb: determining bond info from distances.
Warning: 2GB1.pdb: Determining default bond distances from element types.
注意PDB文件中未包含原子间的键的信息, CPPTRAJ会依照原子间距离和元素类型尝试给出原子之间的键连接信息.
现在读入2GB1分子作为参考结构, 而2GB1无法直接使用第一个拓扑文件, 需要单独指定别的拓扑文件, 类似于参考结构, 拓扑文件也可由序号、名称或者特定标记名来指定.
parmindex <#>: 使用拓扑索引序号<#>.parm <name | tag>: 使用文件名或特定标记名来调用拓扑文件.
> reference 2GB1.pdb parm 2GB1.pdb [GB1]
Reading '2GB1.pdb' as PDB
'2GB1.pdb' is a PDB file, Parm 2GB1.pdb (reading 1 of 1)
接下来是定义rms分析的命令, 与之前不同的是, 首先我们将要分析的trpzip2与GB1链上的残基不完全相同, 还有拓扑中的残基顺序也可能不同, 因此我们只用alpha碳原子进行计算; 其次, 需对这两个不同分子分别做相应的索引来计算其相同原子的RMSD.
> rms ToGB1 ref [GB1] :1-12@CA :42-47,50-55@CA out rmsd3.agr
Mask [:42-47,50-55@CA] corresponds to 12 atoms.
RMSD: (:1-12@CA), reference is reference frame 2GB1.pdb (:42-47,50-55@CA), with fitting.
使用onlyframes指令可让trpzip2轨迹中第一个结构在实现最优重叠的RMSD时输出.
> trajout trpzip2.overlap.mol2 onlyframes 1
Writing 'trpzip2.overlap.mol2' as Mol2
Saving frames 1
输入run进行轨迹处理, 你会发现输出结果中轨迹处理时所选择目标原子的数目与参考结构中对应原子数目是一致的.
ACTION SETUP FOR PARM 'trpzip2.ff10.mbondi.parm7' (1 actions):
0: [rms ToGB1 ref [GB1] :1-12@CA :42-47,50-55@CA out rmsd3.agr]
Mask [:1-12@CA] corresponds to 12 atoms.
处理完之后, 用CPPTRAJ中可能被安装的xmgrace直接查看结果:
> xmgrace rmsd3.agr
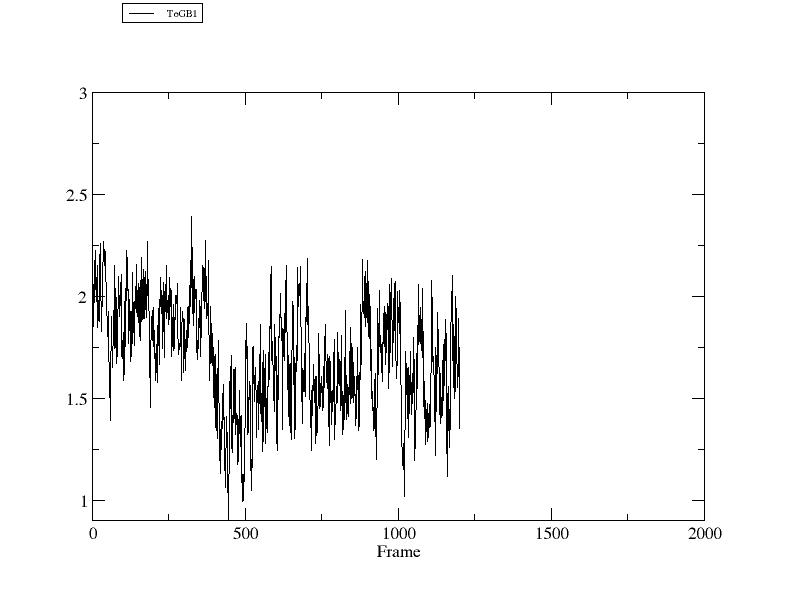
如果查看输出的最优重叠时的文件, 你会发现trpzip2与GB1指定的链已经达到了最好的叠合.
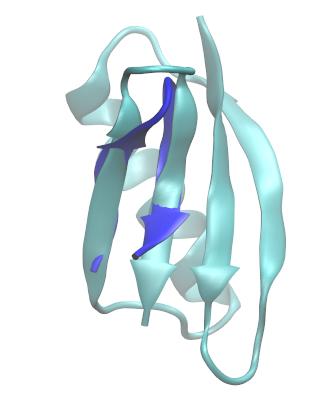
Trpzip2的链(蓝色)与GB1发夹对应的链(青色)相叠合
6. 相关文件
以下由本教程测试输出的文件可用来检验你的模拟结果的正确性.
