- 2018-04-09 20:30:24
问: 有没有办法在GROMACS运行中根据一定的条件改变体系的拓扑文件?
没有, 除非去修改GROMACS的源代码. 目前的大多数MD程序, 其体系的拓扑都是固定的, 是在运行前就决定的, 在整个MD运行过程中都不会改变. 只有这样, 程序才可以方便地进行并行化, 提高性能. 如果在运行时每一步都要重新确定体系的拓扑, 原则上是可行, 但结果就是程序很难并行, 运行速度大大降低.
问: 那如果我 确实 需要这样的功能, 该怎么办呢?
三种解决方法: 换用支持这种做法的程序, 修改GROMACS源代码, 或者采用最简单分段模拟方法.
问: 能说说最简单的分段模拟如何做么?
就是模拟一段时间, 停下来, 改变拓扑后, 使用前一段的最终构型做初始构型继续模拟. 这是最naive和straightforward的方法了. 本质上就是将GROMACS当作一个引擎, 然后写一个外部的脚本或程序来驱动它. 这当然比修改GROMACS源代码简单得多, 而且可以实现任意的修改, 而不单单是改变拓扑. 但这种方法的缺点也很明显, 那就是运行慢.
问: 给个示例说明一下吧. 谢谢.
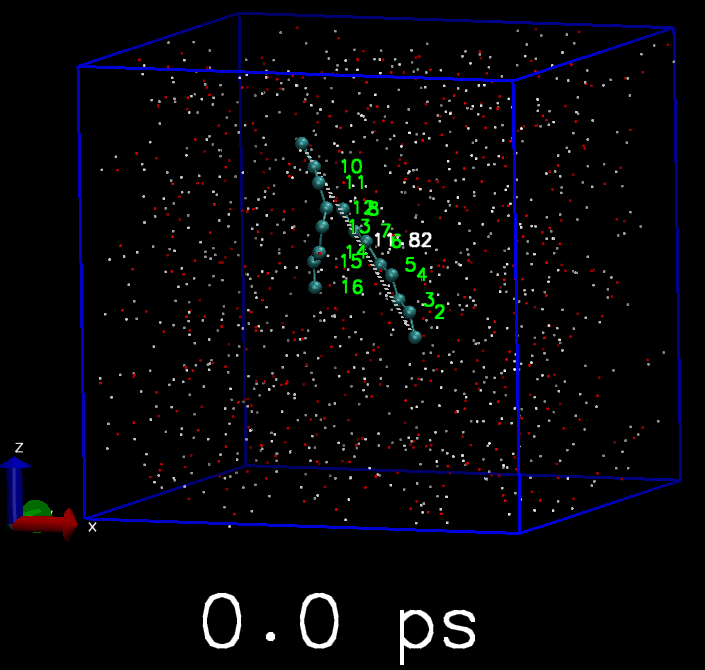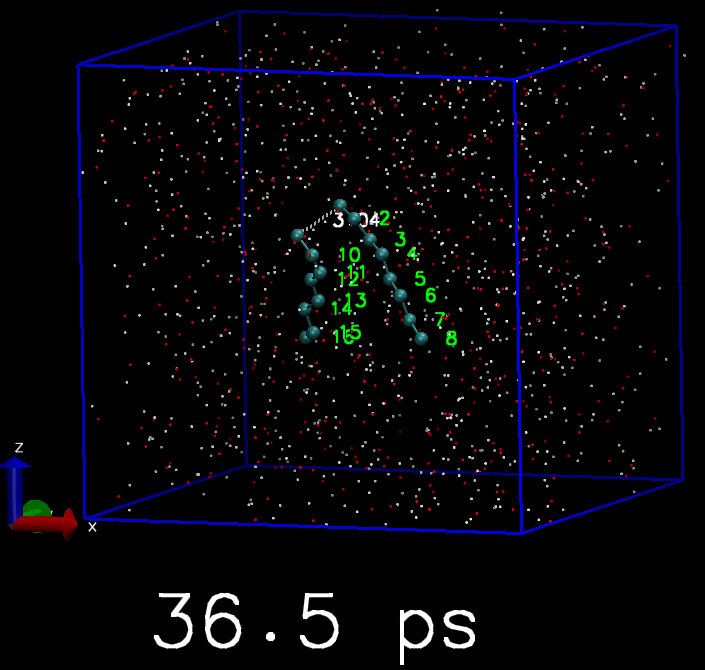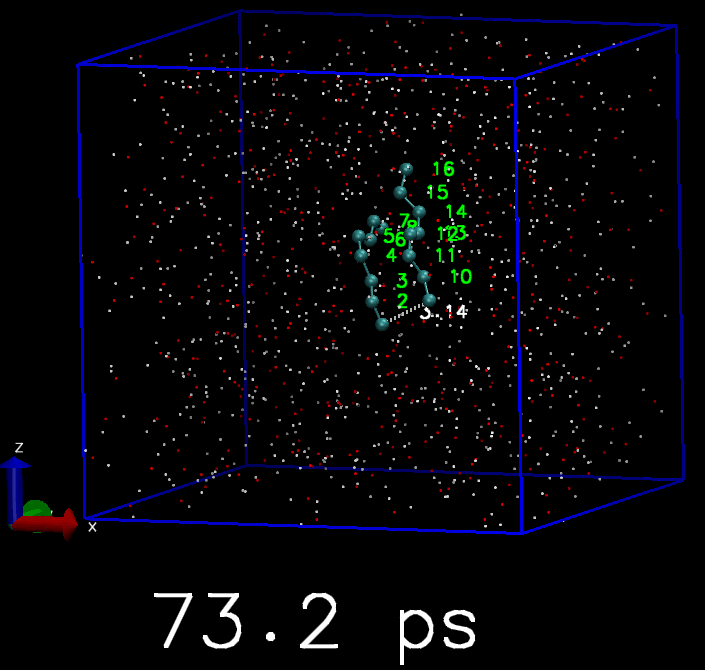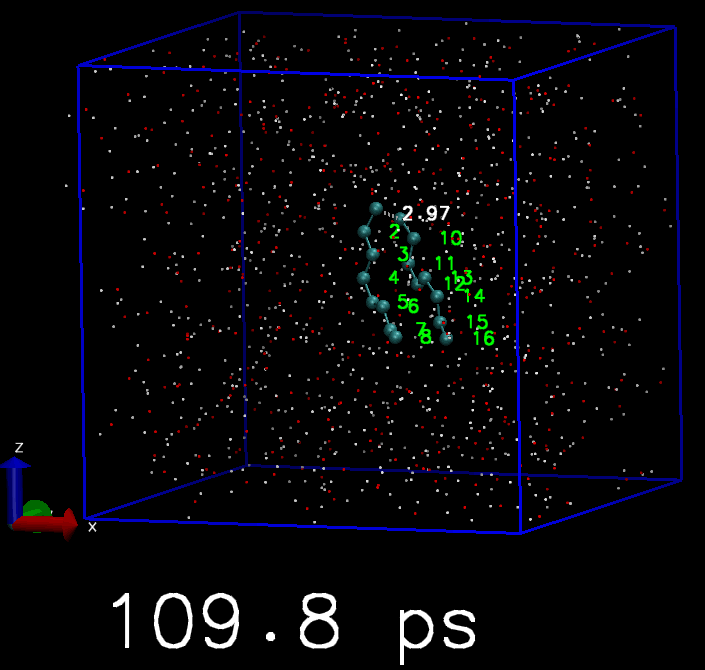
我简单地写了一个脚本, 来驱动GROMACS模拟. 体系是两个C8分子. 开始时这两个C8分子是分离的, 但每隔10 ps, 就会在两个C8分子中对应的原子间添加简谐势函数, 平衡长度为3 埃, 直到所有的C8原子都被简谐势束缚住, 成为类似双链的分子. 下面是模拟的轨迹, 你会看到慢慢地两个C8分子就交联到一起了.