- 2020-03-25 09:48:58
上篇文章中我们整理了VMD中的向量与矩阵操作说明, 这篇文章中我们来具体示例一下如何使用这些函数对分子坐标进行变换, 以创建一些特殊的构型.
我们使用的示例分子如下, xyz格式:
| mol.xyz | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 | 24
mol
C -1.615 -0.739 -3.043
C -0.076 -0.706 -3.045
H -1.963 -1.227 -3.929
H -1.959 -1.275 -2.183
C 0.469 -2.145 -3.087
H 0.268 -0.169 -3.905
H 0.272 -0.218 -2.159
H 1.539 -2.122 -3.089
H 0.126 -2.682 -2.227
H 0.122 -2.633 -3.974
C -2.160 0.701 -3.001
C -3.700 0.667 -2.999
H -1.817 1.237 -3.861
H -1.812 1.188 -2.114
C -4.245 2.107 -2.957
H -4.047 0.179 -3.885
H -4.043 0.131 -2.139
H -3.897 2.594 -2.070
H -3.901 2.643 -3.817
C -5.784 2.073 -2.955
O -6.394 1.131 -3.525
N -6.541 3.141 -2.286
H -7.407 2.773 -1.946
H -6.007 3.500 -1.521 |
如何运行tcl代码
三种方式:
- 将代码复制粘贴到VMD的命令行窗口, 或者
- 将代码保存为
file.tcl, 放到与分子坐标文件相同的路径下, 然后在VMD的命令行窗口中执行source file.tcl, 或者 - 使用VMD的
Extensions -> TkConsole
载入分子, 显示坐标轴和原子序号
打开分子坐标文件mol.xyz, 使用自定义的函数显示坐标轴(showaxis)和原子序号(labelatom), 便于查看原子和操作后的效果.

原点转换
我们首先将分子整体平移, 使N22位于原点.
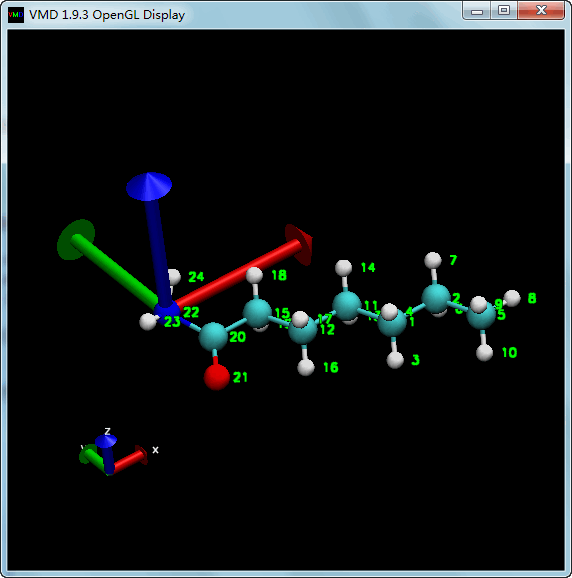
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | # 选中整个分子
set mol [atomselect top all]
# 选中N22
set N [atomselect top "serial 22"]
# 获得N22的坐标
set vN [lindex [$N get {x y z}] 0]
# 设定平移值为N22坐标的负值
set m0 [transoffset [vecinvert $vN]]
# 施加矩阵变换
$mol move $m0 |
上面的操作示例了坐标变换的基本流程, 根据需要获得变换矩阵, 然后对分子施加变换矩阵.
示例中的最后两步操作也可以使用$mol moveby [vecinvert $vN]命令代替, 效果相同. 我们这里使用矩阵操作, 因为使用矩阵便于后面的联合操作.
分子轴平行于坐标轴
接下来, 我们以N22-C5为分子轴, 变换后使得N22-C5平行于x轴
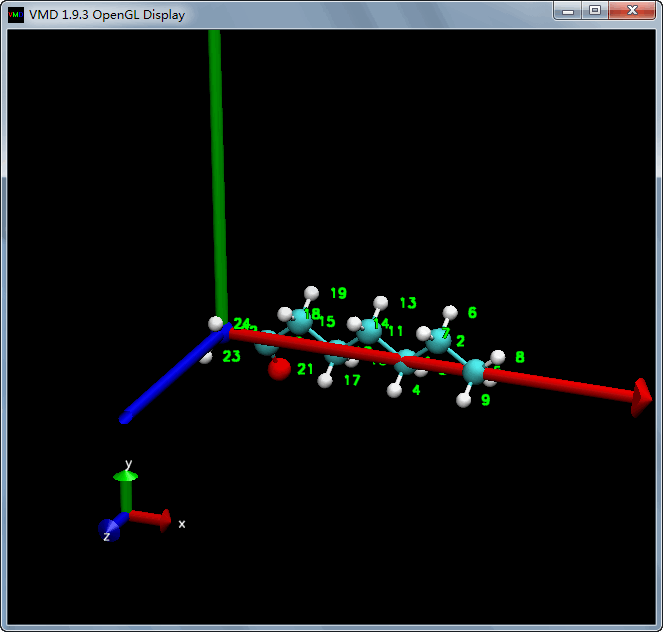
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 | # 选中整个分子
set mol [atomselect top all]
# 选中N22
set N [atomselect top "serial 22"]
# 获得N22的坐标
set vN [lindex [$N get {x y z}] 0]
# 设定矩阵偏移值为N22坐标的负值
set m0 [transoffset [vecinvert $vN]]
# 施加矩阵变换
$mol move $m0
# 选中C5
set C [atomselect top "serial 5"]
# 获得C5的坐标
set vC [lindex [$C get {x y z}] 0]
# 设定分子轴 N-->C
set vNC [vecsub $vC $vN]
# 获得将 N-->C 变换为x轴所需的矩阵
set m1 [transvecinv $vNC]
# 施加矩阵
$mol move $m1 |
上面进行了三次矩阵操作, 每次施加是单独进行的, 这样方便查看效果, 但写起来不方便, 效率也不好. 我们可以先计算出这些矩阵的乘积, 然后只施加一次操作就好了. 值得注意的是矩阵相乘的顺序.
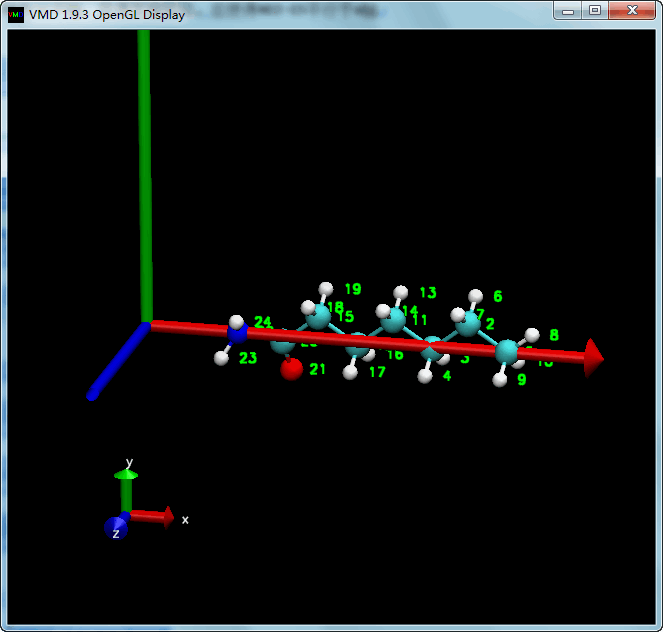
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | # 选中分子
set mol [atomselect top all]
# 选中N22, C5
set N [atomselect top "serial 22"]
set C [atomselect top "serial 5"]
# 获得N22, C5的坐标
set vN [lindex [$N get {x y z}] 0]
set vC [lindex [$C get {x y z}] 0]
# 设定分子轴 N-->C
set vNC [vecsub $vC $vN]
# 设定矩阵偏移值为N22坐标的负值
set m0 [transoffset [vecinvert $vN]]
# 获得将 N-->C 变换为x轴所需的矩阵
set m1 [transvecinv $vNC]
# 沿x轴平移 5A, 以便随后的旋转
set m2 [transoffset {5 0 0}]
# 所有操作, 注意矩阵顺序.
set m [transmult $m2 $m1 $m0]
$mol move $m |
如果你十分清楚自己在做什么, 可以不使用临时矩阵, 直接一步到位.
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 | # 选中分子
set mol [atomselect top all]
# 选中N22, C5
set N [atomselect top "serial 22"]
set C [atomselect top "serial 5"]
# 获得N22, C5的坐标
set vN [lindex [$N get {x y z}] 0]
set vC [lindex [$C get {x y z}] 0]
# 设定分子轴 N-->C
set vNC [vecsub $vC $vN]
# 偏移值为N22坐标的负值
# 将 N-->C 变换为x轴
# 沿x轴平移 5A
$mol move [transmult [transoffset {5 0 0}] [transvecinv $vNC] [transoffset [vecinvert $vN]] ] |
绕轴旋转
在前一步结构的基础上, 我们对分子进行旋转, 旋转时需要确定旋转中心, 旋转轴, 旋转角度. 默认的旋转中心是原点, 旋转轴可以是x, y, z轴, 或任意轴. 我们进行前面的变换就是为了旋转方便, 如果不介意, 可以直接在原坐标的基础上进行旋转, 但那种情况下确定旋转轴会比较麻烦.
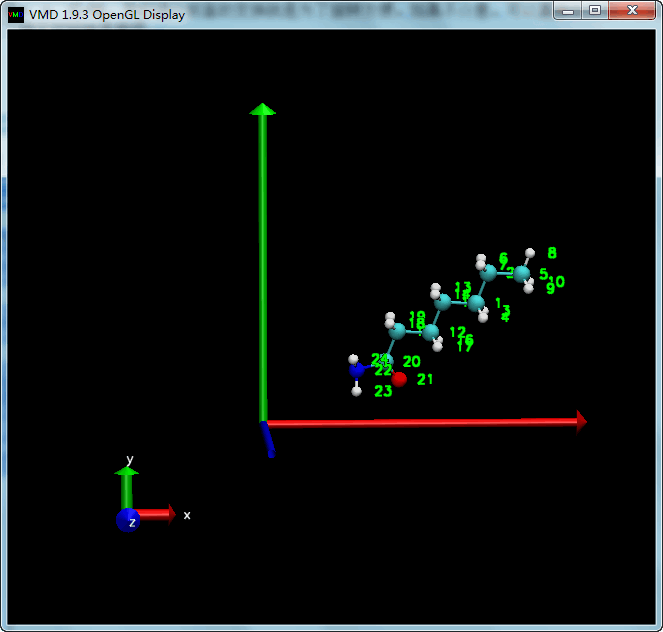
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | # 选中分子
set mol [atomselect top all]
# 选中N22, C5
set N [atomselect top "serial 22"]
set C [atomselect top "serial 5"]
# 获得N22, C5的坐标
set vN [lindex [$N get {x y z}] 0]
set vC [lindex [$C get {x y z}] 0]
# 设定分子轴 N-->C
set vNC [vecsub $vC $vN]
# 偏移值为N22坐标的负值
# 将 N-->C 变换为x轴
# 沿x轴平移 5A
# 绕z轴旋转30度
$mol move [transmult [transaxis z 30] [transoffset {5 0 0}] [transvecinv $vNC] [transoffset [vecinvert $vN]] ] |
多次旋转
利用循环, 我们可以进行多次旋转, 同时保存每次旋转后的构型. 注意循环添加的位置.
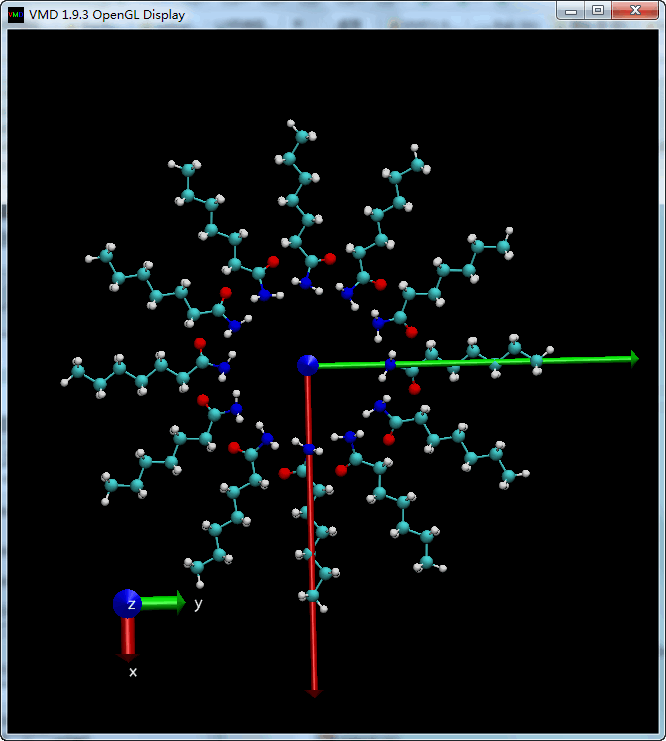
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 | # 选中分子
set mol [atomselect top all]
# 选中N22, C5
set N [atomselect top "serial 22"]
set C [atomselect top "serial 5"]
# 旋转, 每次30度
for {set t 0} {$t <= 360} {incr t 30} {
# 获得N22, C5的坐标, 分子轴 N-->C
set vN [lindex [$N get {x y z}] 0]
set vC [lindex [$C get {x y z}] 0]
set vNC [vecsub $vC $vN]
# 偏移值为N22坐标的负值
# 将 N-->C 变换为x轴
# 沿x轴平移 5A
# 绕z轴旋转 t
$mol move [transmult [transaxis z $t] [transoffset {5 0 0}] [transvecinv $vNC] [transoffset [vecinvert $vN]] ]
# 保存xyz文件
$mol writexyz "mol_$t.xyz"
} |
我们可以得到的所有xyz文件合并到一起, cat mol_*.xyz >mol~.xyz, 除去不需要的原子数目行和标题行, 并修改总原子数就可以使用了.
旋转加平移, 螺旋构型
一边旋转, 一边平移, 就是螺旋了.
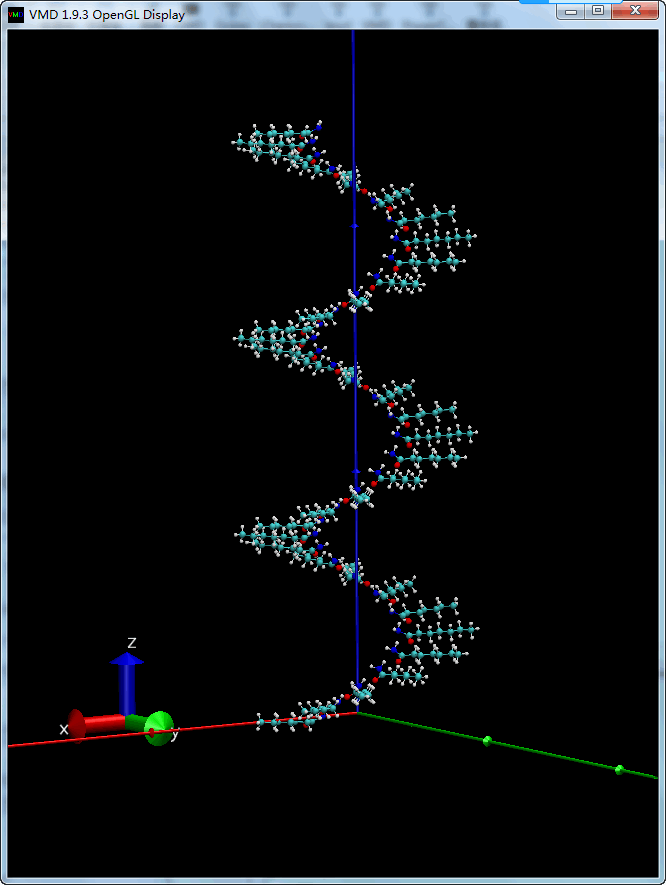
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 | # 选中分子
set mol [atomselect top all]
# 选中N22, C5
set N [atomselect top "serial 22"]
set C [atomselect top "serial 5"]
# 旋转, 每次30度
set dt 30
set n [expr 3*360/$dt]
for {set i 0} {$i <= $n} {incr i} {
# 获得N22, C5的坐标, 分子轴 N-->C
set vN [lindex [$N get {x y z}] 0]
set vC [lindex [$C get {x y z}] 0]
set vNC [vecsub $vC $vN]
# 偏移值为N22坐标的负值
# 将 N-->C 变换为x轴
# 沿x轴平移 5A
# 绕z轴旋转 t
# 沿z轴平移 z, 注意使用方式
set t [expr $i*$dt]
set z [expr $i*2]
$mol move [transmult [transoffset "0 0 $z"] [transaxis z $t] [transoffset {5 0 0}] [transvecinv $vNC] [transoffset [vecinvert $vN]] ]
# 保存为xyz文件
$mol writexyz "mol_$z-$t.xyz"
} |
旋转, 平移, 圆柱构型
如果平移和旋转相互独立, 得到圆柱构型.
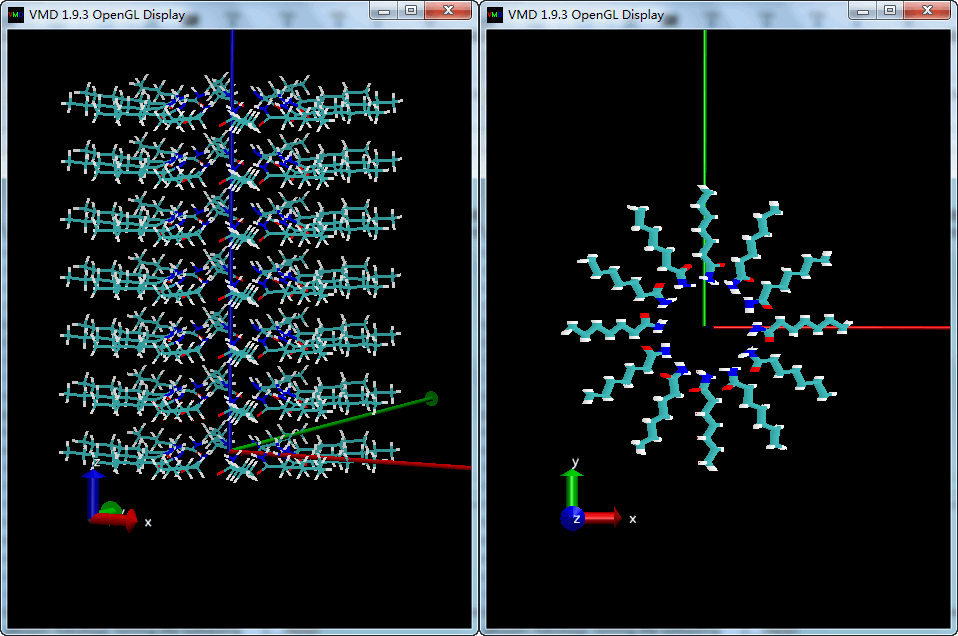
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 | # 选中分子
set mol [atomselect top all]
# 选中N22, C5
set N [atomselect top "serial 22"]
set C [atomselect top "serial 5"]
# 旋转, 每次30度
set dt 30
set n [expr 3*360/$dt]
for {set z 0} {$z <= 30} {incr z 5} {
for {set i 0} {$i <= $n} {incr i } {
# 获得N22, C5的坐标, 分子轴 N-->C
set vN [lindex [$N get {x y z}] 0]
set vC [lindex [$C get {x y z}] 0]
set vNC [vecsub $vC $vN]
# 偏移值为N22坐标的负值
# 将 N-->C 变换为x轴
# 沿x轴平移 5A
# 绕z轴旋转 t
# 沿z轴平移 z, 注意使用方式
set t [expr $i*$dt]
$mol move [transmult [transoffset "0 0 $z"] [transaxis z $t] [transoffset {5 0 0}] [transvecinv $vNC] [transoffset [vecinvert $vN]] ]
# 保存为xyz文件
$mol writexyz "mol_$z-$t.xyz"
} } |
总结
使用VMD的矩阵操作进行建模, 总体而言比自己写代码方便, 但tcl语言 太 差 了, 坑很多, 需要熟悉, 熟悉了之后也会很快忘记, 每次用的时候都要查语法. 详细的语法可以参考我整理的TCL培训教程.
