- 2022-02-09 22:26:57
我的gmx_mmpbsa脚本大致是我用来学习MM-PBSA的原理和计算流程的, 虽然也有人将它用于实际计算. 既然有人在用, 无论学习还是处理自己的体系, 于我都是额外的收益. 既有益处, 也就无可避免地有坏处, 那就是有人会发现脚本中存在bug, 促我不断地更新.
最近就有人发现了脚本中的bug, 主要涉及PBSA计算时所用的原子半径不一致, 特别是对于非蛋白分子. 我检查了一下, 发现原来脚本所用的原子半径是根据PDB中的原子名称确定的. 对蛋白而言, 原子名称大致对应于原子类型, 所以问题不大, 但对于非蛋白的小分子配体, 基本没有通用的原子名称, 所以无法根据原子名称关联原子类型, 也就无法正确确定原子半径了. 对氢原子, 这个问题最为突出, 因为氢原子的原子半径一般是与其所连原子的种类有关的, 很难从其名称中推断出来.
在修正这个bug的时候, 顺便检查了一下计算PBSA时到底该用哪套原子半径. 文献上讨论很多, 恕我能力精力有限, 无法一一确认, 就直接看AMBER的处理了. 最新AMBER 20手册787页的表35.4总结了各种情况:
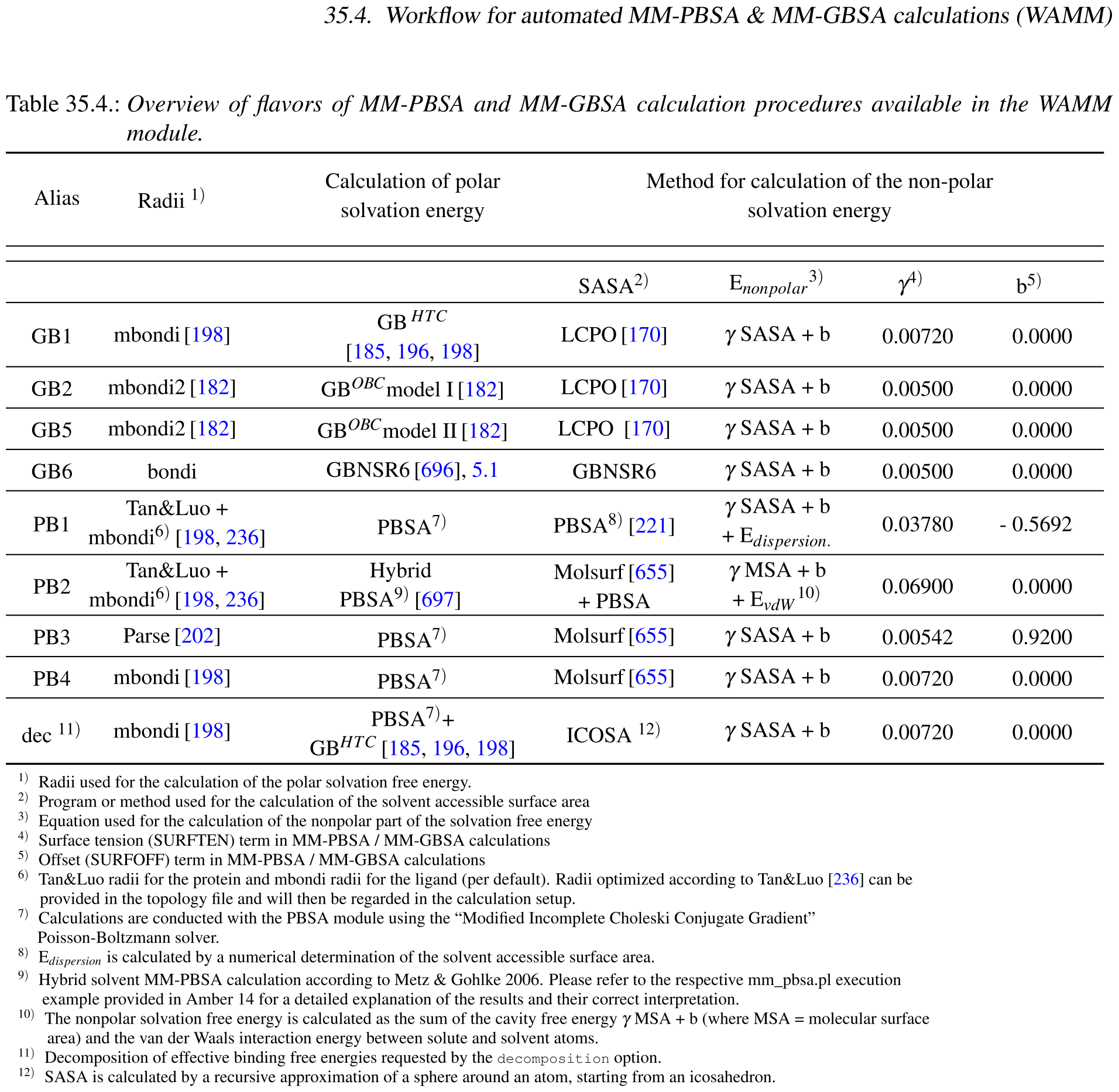
根据这个表中所用半径以及非极性项的处理, 只有其中的PB4是适合gmx_mmpbsa脚本的, 其他的或者确定半径的方法不好实现, 或者非极性项不好计算, 所以我就将脚本的PBSA处理方法改成与AMBER的PB4一致了. PB4方案所用的原子半径为mBondi, 具体的数值可以查看AmberTools源码中的parmed/tools/changeradii.py脚本. 这种修改肯定会对计算结果产生影响, 我没有实测, 但猜测影响比较小.
既然已经花了不少时间重新熟悉脚本, 并更新了脚本, 那顺便也将以前写下的待办项也完成下吧. 首要的待办项是计算丙氨酸扫描CAS, 也就是将某些蛋白残基变为丙氨酸后再计算MM-PBSA. 这个事情并不复杂, 但我以前的想法是要完成一个通用的方案, 实现所有氨基酸彼此之间都可以相互突变, 而不是只能突变为丙氨酸. 但这个功能还是有点难弄, 要写不少代码, 所以我就拖下来了. 现在既然已经暂时放弃了更通用的方案, 只实现一个CAS还是简单的. 查看了一遍所有残基的结构, 发现除甘氨酸GLY, 脯氨酸PRO, 成二硫键的半胱氨酸CYX外, 要突变为丙氨酸ALA, 只要将原残基的几个原子改为相应的氢原子, 并删除其他无关原子即可. 当然, 氢原子的位置要做相应的调整, 突变后的原子类型也要与丙氨酸保持一致.
夜已经深了,我合上笔记本,黯淡了灯光,望望窗外,雪白的大地泛着茫茫。这是寒冬的夜, 没有行人。我对自己说道:“关灯吧!你还没读够?”他则说:“再等一会。我这就读完哲科文推送的《gmx_mmpbsa更新: 修正原子半径bug, 改用AMBRR PB4, 丙氨酸扫描CAS》了。”
