- 2023-06-02 00:16:15
在建模或创建拓扑的时候, 经常会遇到需要调整分子中原子顺序的烦心事. 可以想见的情形, 可以预见的用途就不多说了. 我以前也有过总结, 还写了一个在线工具, 可惜使用并不是很方便, 也就没分享出来. 所以遇到这样的问题时, 如果并不需要经常做, 我一般就建议手动调整, 原子数目少的时候也并不费事. 但如果原子数目多起来了, 且排列没有规律, 手动调整并非不可行, 只是很繁琐, 且容易出错. 即便存在这些缺点, 手动调整仍是第一位的. 手动操作不仅让你 真 正 知道需要做的是什么, 更能让你在工具出错时, 知道如何补救.
在自己手动调整过一些实例, 了解了具体如何操作之后, 我们就可以试着将繁琐的手动操作程序化/自动化. 这可以借助一些脚本/程序, 甚至是AI机器人. 省出的时间可以让我们思考一些其他的问题, 无论人生的还是研究的, 或者只是单纯享用这些时间. 为此, 我就再次考虑了一下这个问题, 查阅了一下最近的资料, 改进了一下以前的一个半成品脚本, 算是给出一个稍微完整的解决方案.
从理论方面讲, 分子匹配算是子图同构问题, 并不存在什么高效算法, 所有算法都不过是尽可能的多利用已知信息, 减少需要匹配的数目. 所以这里没有银弹, 也不存在黑狗血, 只有简单粗暴, 逐个比较. 关于实现算法, 可以参考使用 VF2 算法进行子结构搜索, 里面有对VF2算法及其源码的分析.
我暂时没有时间/能力自己实现VF2算法, 所以就先基于NetworkX实现了所需的功能. 虽然我一直不是很喜欢Python, 但似乎无论什么领域都能看到它的影子, 没奈何. 因为Python脚本使用起来并不方便, 所以我就将其打包成一个exe, 但因此也就只能在Windows下使用.
程序
说明
matchmol脚本用于匹配两个分子(参考分子, 待匹配分子)中原子的编号顺序, 两个分子的构型可以相同, 也可以不同. 匹配只基于每个原子的原子类型(图的顶点属性)和原子间的成键类型(图的边属性), 而不会考虑具体的键长键角等几何信息, 也就是只考虑分子的拓扑结构. 匹配后还会计算两个分子的RMSD, 用以表征分子空间结构的差距.
脚本首先读入分子结构文件, 格式支持pdb或mol2. 建议使用mol2, 因为其中可以指定更精细的原子类型和成键类型, 从而更容易匹配. 但需要注意原子类型和成键类型的指定在两个分子要相同, 否则可能出现无法匹配的情况. 如果可能, 可以使用AMBER的GAFF工具处理一下, 生成包含具体原子类型和成键类型的mol2文件. 使用pdb的话, 文件中必须使用CONECT指定成键情况. 匹配时原子类型只采用元素符号, 成键类型也只有一种, 所以效率不高.
读入分子后, 根据分子的成键情况将其转换为图, 顶点为原子, 属性为原子类型, 边为键, 属性为成键类型. 然后借助NetworkX的isomorphism.GraphMatcher函数, 对两个分子的图进行匹配, 得到所有原子的匹配信息.
对绝大多数分子, 氢原子都处于终端位置, 所以同一重原子所连的多个氢原子在匹配时无法区分, 可以有多种匹配顺序. 一般情况下这不是大问题. 为了进一步精化氢原子的匹配, 脚本会根据得到的所有原子匹配信息, 使用重原子进行叠合, 并计算叠合重原子后重原子以及所有原子的RMSD. 再根据重原子叠合后氢原子的位置构建氢原子匹配的误差矩阵, 借助NetworkX的max_weight_matching函数单独对氢原子进行匹配. 重新调整氢原子匹配顺序后, 会计算所有原子叠合后的RMSD. 一般情况下, 这次得到的所有原子的RMSD会小于前一次的, 因为对氢原子顺序进行了调整. 需要说明的是, 这种处理氢原子的方式虽然可以进一步减小RMSD, 但对于顺反异构, 手性等氢原子顺序敏感的情况, 并不能保证匹配一定正确, 所以在这种情况下, 最终仍需要人工检查.
叠合并计算RMSD使用的是Kabsch算法。
除屏幕输出外, 脚本还会输出一个pdb文件, 文件名称为待匹配分子的文件名称~match.pdb, 其中包含4套坐标:
- 参考分子的原始坐标
- 待匹配分子经重原子叠合后的坐标(以全原子匹配后的顺序)
- 待匹配分子经所有原子叠合后的坐标(以调整氢原子后的顺序)
- 待匹配分子的原始坐标(以调整氢原子后的顺序)
前3套坐标用于检查匹配结果, 最后一套坐标用于需要使用带匹配分子调整顺序后的原始坐标的情况.
为了方便检查匹配结果, 脚本支持-t选项, 开启后输出文件中的的两套叠合坐标会进行平移, 以便查看时错开.
建议借助Jmol核查匹配结果, 可以将前三套坐标同时显示在屏幕上, 并可以显示每套坐标调整后的编号, 或原始编号.
示例
使用自带的几个分子.
简单小分子: ref-1.pdb和mol-1.pdb
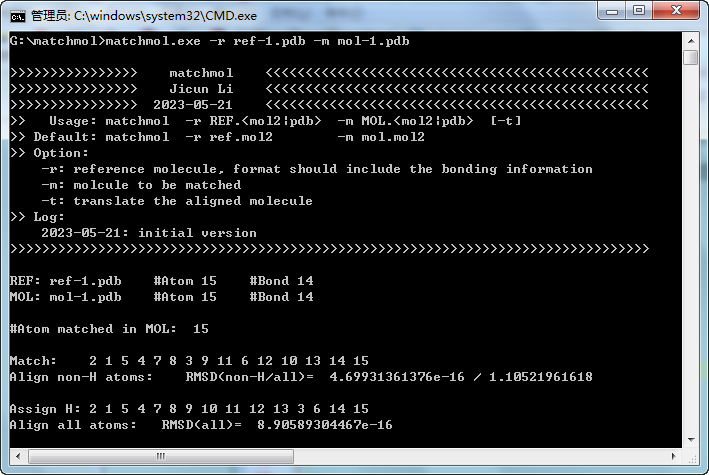
这两个分子除原子顺序不一致外, 构型相同. RMSD结果证实了这一点. Jmol显示后, 4套坐标完全重合, 调整氢原子后原子编号一致.
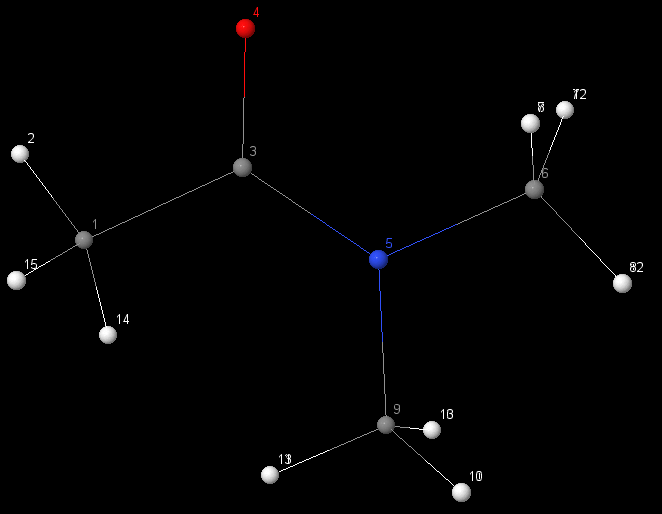
稍复杂分子: ref-2.mol2和mol-2.mol2
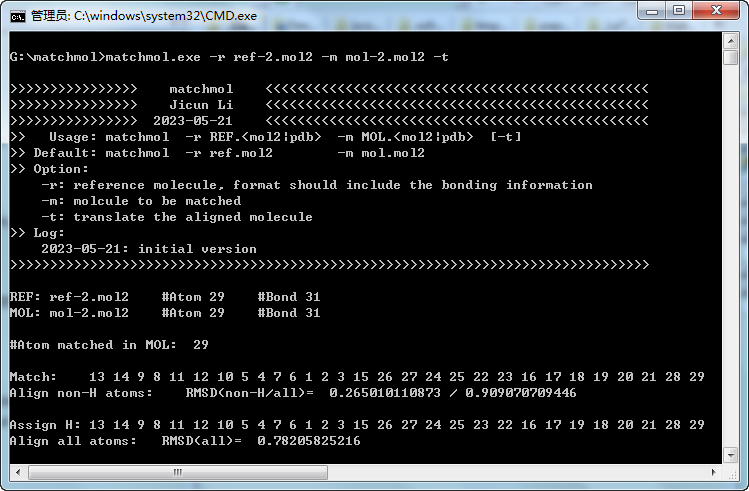
这两个分子的区别主要在于一个六元环发生了扭曲. 未调整氢原子时, 有几个氢原子的匹配并不准确.
指定-t选项获得输出文件后, 使用Jmol加载前三套坐标, 显示原子编号或原子名称, 可以比较详细地核查匹配结果.
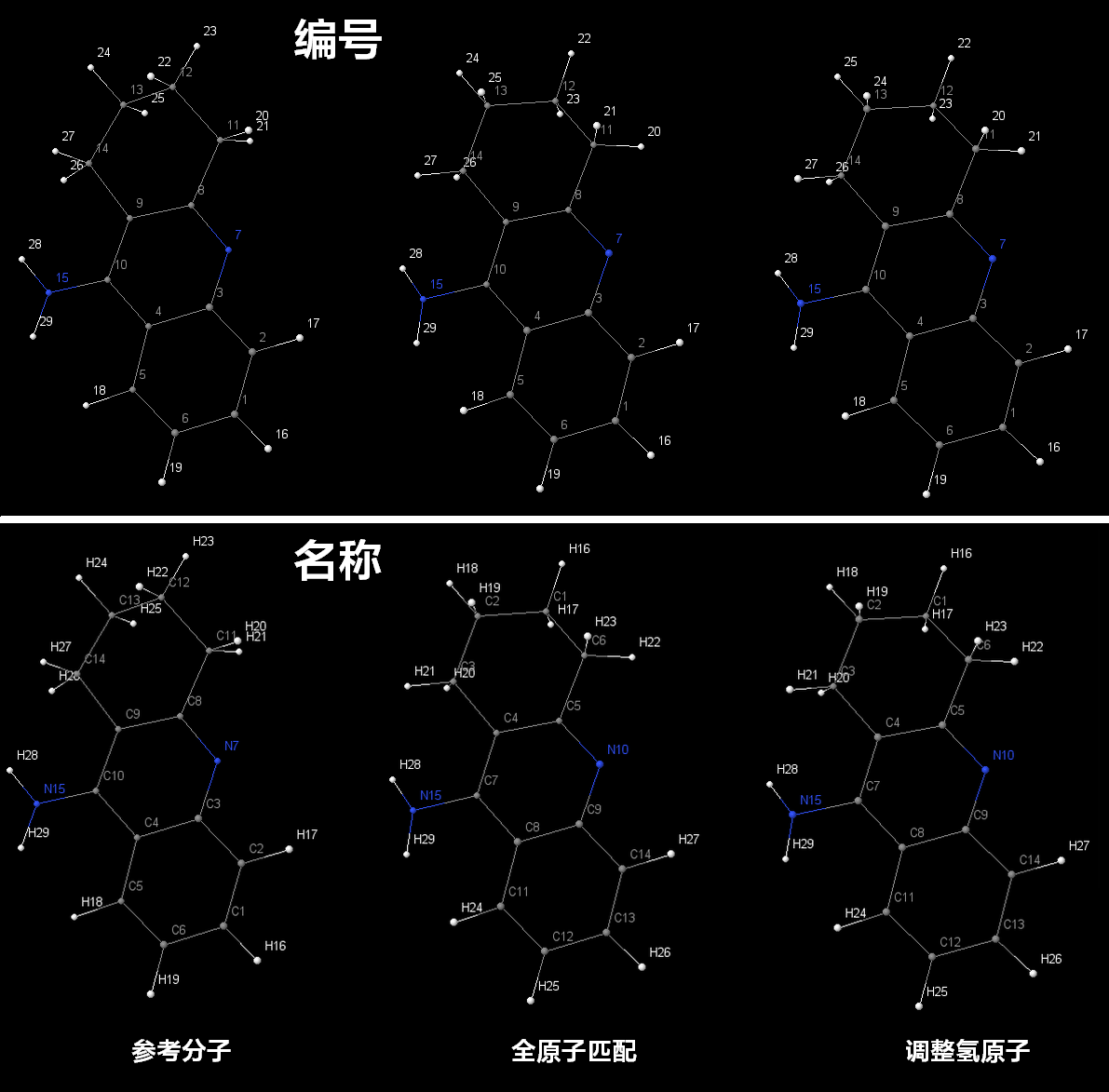
多糖分子: ref-3.pdb和mol-3.pdb
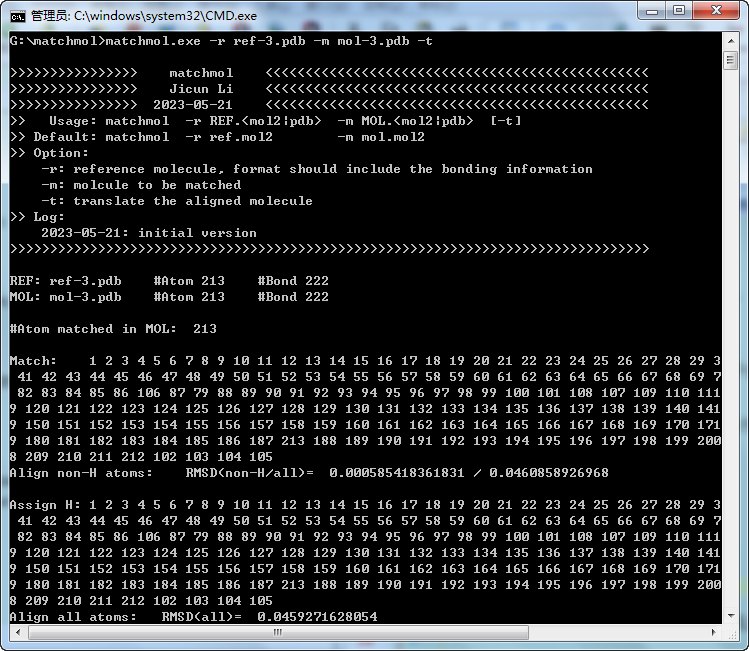
不同来源的两个淀粉螺旋链, 手动匹配繁琐且易出错.
待改进
- 位置敏感氢原子的处理
- 基于js的在线工具
