- 2015-12-10 21:03:38 本文译自All-atom automatic OPLS-AA topology generator, 为TPPMKTOP的说明文档.
- 2017-07-11 08:37:00 补充两个常见问题
- 2018-04-24 13:43:39 修正错误
生成OPLS-AA力场的拓扑文件非常复杂, 因为此力场包含的原子类型非常多, 超过800种. 但即便使用如此多的原子类型, OPLS-AA力场仍不可能描述所有分子的化学结构. 因此, 当文献中给出了新化学片段的参数后, OPLS-AA力场的原子类型也会随之增加. TPPMKTOP是一个自动化的工具, 可用于生成OPLS-AA力场的拓扑文件. 它提供了免费的网络服务, 网址为http://erg.biophys.msu.ru/tpp. TPPMKTOP使用了MySQL数据库, 其中包含了有关原子类型指认的力场参数和信息. 这个数据库由我们研究组负责持续升级. TPPMKTOP是一个开源项目, 你可以联系comconadin@gmail.com以便获得最新版本.
下面我们介绍一下TPPMKTOP所用的原子类型指认算法, 并以下面的分子为例说明一下TPPMKTOP的原理.
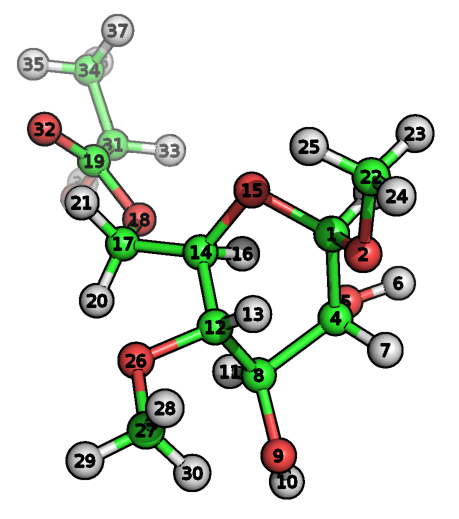
其PDB文件可在这里下载.
当将PDB文件上传到服务器后, 会执行下面的命令:
tppmktop -i file.pdb -f OPLS-AA -o file.itp -r file.rtp -v -n -l lack.itp
其中, file.pdb为你上传的PDB文件, OPLS-AA为力场名称. 上面的命令执行成功后程序会给出3个输出文件: file.itp为可单独使用的拓扑文件, file.rtp为可用于pdb2gmx的拓扑文件, lack.itp文件中定义了缺失的力场参数. 此外, 还会给出所有的输出文件, 日志文件和控制台输出. 让我们以上面的分子为例来说明生成拓扑的细节.
处理步骤
第一步: 程序输出内部统计的结果
Input file format: Protein Data Bank.
Forcefield OPLS-AA was found in database.
Description: Optimized Potential for Liquid Simulation. All-atomic variant..
Total statistics:
865 atoms, 314 bonds, 988 angles,
1269 dihedrals, 0 nonbonded parameters.
在控制台输出中, TPPMKTOP会打印出服务器数据库的统计信息, 这些值对应于所选力场在数据库中的总的原子类型数, 键合相互作用参数的数目.
第二步: 读入并处理输入的化学结构
控制台输出
Atoms: [.......................................]
日志文件中会输出下列信息
Trying to read structure from 'file.pdb'.
LIG - C: 1(1) [ -1.409, 2.132, -0.108] QMname: C
LIG - O: 2(2) [ -2.614, 2.502, -0.908] QMname: O
LIG - H: 3(3) [ -0.928, 3.050, 0.331] QMname: H
LIG - C: 4(4) [ -2.042, 1.200, 1.158] QMname: C
...
从这些输出你可以检查原子名称, 坐标, 元素名称(非常重要!), 检查TPPMKTOP是否处理正确.
第三步: 构建原子间的共价连接矩阵
原子间的共价连接矩阵表示哪些原子之间有共价键相连, 但这不会在日志中输出. 这一步是通过调用openbabel外部过程完成的. 你可以通过将PDB文件转换为SMILES格式来检查这一步是否正确完成了.
第四步: SMARTS匹配
构建完共价连接矩阵后, 会启动SMARTS匹配过程, 会在输出中显示
3 queries proceeded on database
Calculating scores for every atom.. finished!
Patterns are loading. Please wait.. finished.
Starting SMART-fit.
Patterns checked: 296.
同时在日志中会列出所有匹配的SMARTS模式
Starting curious SMART-fitting procedure.
Loading patterns from database...OK!
[OB] Process PAT: [H,C]C(=O)OC having 5 atoms.
[OB] Process PAT: ClC(Cl)(Cl)Cl having 5 atoms.
[OB] Process PAT: Cl[CX4;$(C(Cl)(Cl)(Cl)Cl)] having 2 atoms.
[OB] Process PAT: [+NH4] having 1 atoms.
...
TPPMKTOP会试着将数据库中的每一个模式与分子的化学结构进行匹配, 构建出与每个原子匹配的原子类型, 并进行排序.
第五步: 选择每个原子的原子类型
根据条件得分优先级, SMARTS匹配过程为每个原子选择一个原子类型
Starting atom_alig..
Filling map..
Applying scores...
为理解这一过程的细节, 让我们来看下核心数据库. TPPMKTOP处理完我们的分子后, 在最终的itp文件中, 1号碳原子指认的原子类型为acetal opls_193(缩醛). 为什么? 在SMARTS数据库中, 有两个记录与原子1的化学环境匹配, 第一个为(绿色)
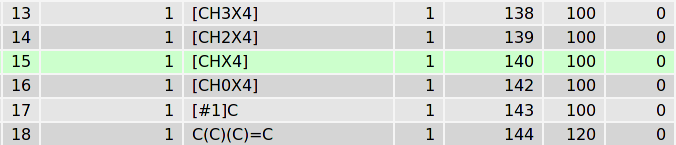
[CHX4]对应于脂肪碳原子, 只含一个氢, 且为4价. 第二个匹配模式(绿色)为

这一记录意味着C-O-[CH](C)-O模式的第三个原子应定义为196号原子类型. 这一SMARTS模式对应于下面这种片段: 一个脂肪碳原子一端与氧相连, 另一端依次连着只含一个氢的脂肪碳原子, 连接了两个碳的氧原子, 一个脂肪碳原子. 选择是基于两个匹配模式的条件得分优先级(越大越好): 196号原子类型得分150, 140号得分只有100. 因此, 程序会选择196号原子类型. 在数据库内部的记录中, 196号原子类型对应于opls_193.
第六步: 划分电荷组
在这一步中, 会根据数据库以及不当二面角(保持平面性或手性)将原子划分为电荷组, 其算法类似于上面的SMARTS匹配.
CHARGEGROUP patterns are loading. Please wait.. finished.
Starting SMART-fit.
Patterns checked: 8..........
Renumbering CGNR according to human-readable style..finished.
IMPROPER patterns are loading. Please wait.. finished.
Starting SMART-fit.
Patterns checked: 2.
第七步: 生成1-4相互作用
如果所选力场需要显式地给出1-4相互作用, 则会将它们自动添加到[ pairs ]部分
Generating 1-4 pairs for FF needs..ok.
第八步: 处理缺失力场参数
缺失的力场参数会写到lack.itp文件的#define部分, 其系数为零. 通过将参数补充完整, 并将其内容复制到主itp文件的开始部分, 你可以很容易地得到完整的拓扑文件. 对我们的例子而言, 没有缺失键合参数.
TPP will write 0 lack parameters to lack.itp.
第九步: 打印电荷
在最后一步, TPPMKTOP会打印出体系的总电荷. 如果为体系中的所有原子都正确地指认了原子类型, 那么这一电荷会等于(或接近)分子的总电荷. 如果仍不相等, 你可以手动修改部分电荷.
Please, correct your charges according to sum: 0.000.
TPPMKTOP finished normally!
你应该检查一下, 直观看来, 指认的原子电荷是否与整个化学结构符合. 指认的所有原子类型会在分号后的注释中列出.
[ atoms ]
1 opls_193 1 LIG C 1 0.300 12.011000 ; C(HCO2): acetal OCHRO
2 opls_186 1 LIG O 2 -0.400 15.999400 ; O: acetal ether
3 opls_194 1 LIG H 1 0.100 1.008000 ; H(CHO2): acetal OCHRO
4 opls_158 1 LIG C 3 0.205 12.011000 ; all-atom C: CH, alcohols
5 opls_169 1 LIG O 4 -0.700 15.999400 ; O: diols
6 opls_170 1 LIG H 5 0.435 1.008000 ; H(O): diols
7 opls_140 1 LIG H 3 0.060 1.008000 ; alkane H.
8 opls_158 1 LIG C 6 0.205 12.011000 ; all-atom C: CH, alcohols
9 opls_169 1 LIG O 7 -0.700 15.999400 ; O: diols
10 opls_170 1 LIG H 8 0.435 1.008000 ; H(O): diols
11 opls_140 1 LIG H 6 0.060 1.008000 ; alkane H.
12 opls_183 1 LIG C 9 0.170 12.011000 ; C(HOR): i-Pr ether, allose
13 opls_185 1 LIG H 9 0.030 1.008000 ; H(COR): alpha H ether
14 opls_183 1 LIG C 10 0.170 12.011000 ; C(HOR): i-Pr ether, allose
15 opls_186 1 LIG O 11 -0.400 15.999400 ; O: acetal ether
16 opls_185 1 LIG H 10 0.030 1.008000 ; H(COR): alpha H ether
17 opls_490 1 LIG C 12 0.190 12.011000 ; C(H2OS) ethyl ester
18 opls_467 1 LIG O 13 -0.330 15.999400 ; AA -OR: ester
19 opls_465 1 LIG C 13 0.510 12.011000 ; AA C: esters - for R on C=O, use #280-#282
20 opls_469 1 LIG H 12 0.030 1.008000 ; methoxy Hs in ester
...
常见问题
引用扩充的力场文件
PPMKTOP扩展了OPLSAA的原子类型, 因此, 有时其给出的拓扑文件中会包含有自定义的原子类型, 如果直接使用的话, grompp时找不到所需的原子类型, 导致出错. 解决方法是下载TPPMKTOP扩充的OPLSAA力场. 下载解压后将其放于类似C:\GMX\GMX5.1.4\share\gromacs\top的目录下, 并将文件夹更名为oplsaa-erg.ff, 在拓扑文件中#include "oplsaa-erg.ff/forcefield.itp"就可以引用新的力场文件了.
异常二面角参数的错误
对于OPLSAA力场的C:\GMX\GMX5.1.4\share\gromacs\top\oplsaa.ff\ffbonded.itp文件, 无论是GROMACS自带的, 还是TPPMKTOP扩充后的, 都存在异常二面角参数对应的[ dihedraltypes ]段. 原始内容如下
[ dihedraltypes ]
; Improper OPLS dihedrals to keep groups planar.
; (OPLS doesnt use impropers for chiral atoms).
; Since these functions are periodic of the form 1-cos(2*x), they are actually
; implemented as proper dihedrals [1+cos(2*x+180)] for the moment,
; to keep things compatible.
; The defines are used in ffoplsaa.rtp or directly in your .top file.
; O?-C -X -Y improper torsion. C can be C_2 or C_3 too.
#define improper_O_C_X_Y 180.0 43.93200 2
; X-NO-ON-NO improper torsion.
#define improper_X_NO_ON_NO 180.0 43.93200 2
; N2-X-N2-N2 improper torsion.
#define improper_N2_X_N2_N2 180.0 43.93200 2
; Z -N?-X -Y improper torsion
#define improper_Z_N_X_Y 180.0 4.18400 2
; Z -CM-X -Y improper torsion. CM can be C= too.
#define improper_Z_CM_X_Y 180.0 62.76000 2
; Z -CA-X -Y improper torsion. CA is any ring carbon (CA,CB,CN,CV,CW,CR,CK,CQ,CS,C*)
#define improper_Z_CA_X_Y 180.0 4.60240 2
这里定义了一些异常二面角类型的势能参数, 但没有给出函数类型参数, 因此如果在拓扑文件中直接使用1 2 3 4 improper_O_C_X_Y这样的形式来定义异常二面角, grompp时就会出错, 给出invalid dihedral type 180的错误. 根据GROMACS手册异常二面角的说明, 这些异常二面角的函数类型应该为4(也可以使用1, 二者没有区别), 所有只要在拓扑文件中所有类似i j k l improper_O_C_X_Y的地方增加函数类型, 改为i j k l 4 improper_O_C_X_Y或i j k l 1 improper_O_C_X_Y即可. 不建议直接修改原始的力场文件, 因为这些参数定义会用于氨基酸的二面角, 修改原始力场文件后会导致pdb2gmx生成的蛋白拓扑文件错误.
以前的做法grompp虽然可以成功, 但生成的拓扑文件是错误的.
改正方法也很简单, 将上面的#define部分中的参数顺序调整与GROMACS需要的顺序一致, 也就是将最后一个表征类型的数字2转移放到平衡角度值180.0前面
