- 2015-12-08 22:16:10 初稿
- 2016-03-28 20:18:18 补注: 不同版本ACPYPE生成的二面角函数类型
- 下载本文用到的文件
使用AMBER的GAFF力场处理有机小分子有很大的优势, 可惜的是GROMACS没有自带GAFF力场, 所以需要组合使用各种软件组合来实现. 这里我们使用的是AmberTools和ACPYPE.
要想使用AmberTools和ACPYPE创建小分子的GAFF力场, 需要先安装这两个工具. 但Windows下AmberTools的安装并不容易. 在这里我提供一个已经编译好的AmberTools+ACPYPE, 它来自Chimera中所带的amber14, 并增加了我自己编译好的RESP程序与ACPYPE程序, 因为Chimera中并没有包含这两个程序. 具体的整合过程请参看Windows下的AmberTools+RESP+ACPYPE.
点击这里下载AmberTools+ACPYPE, 下载后解压到某一目录(路径中不要包含中文字符), 然后新建环境变量AMBERHOME并将其设置为amber14的路径即可.
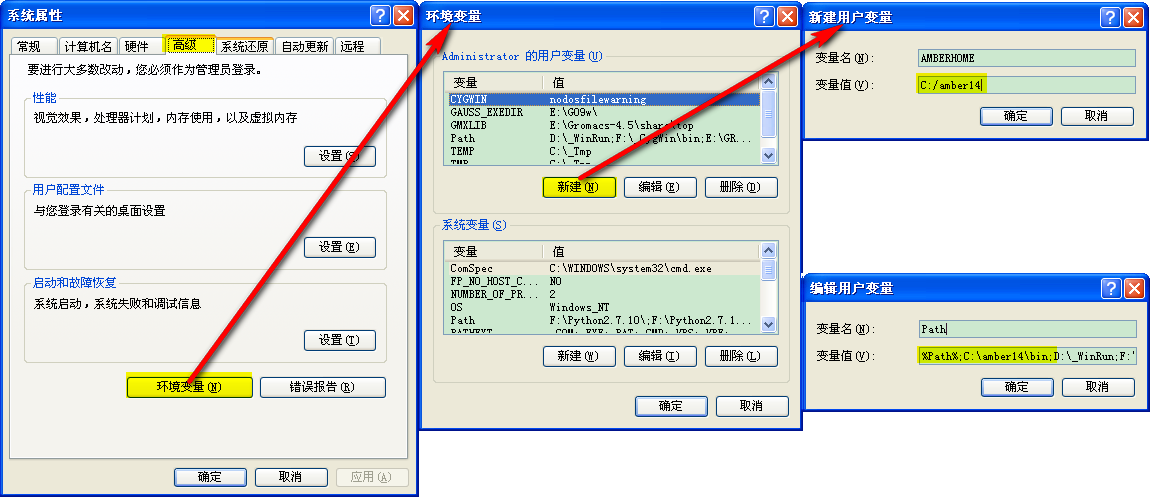
具体操作如下:
右键我的电脑->属性->高级->环境变量
在用户变量中新建AMBERHOME, 并其值设为amber14的路径, 如C:/amber14. 注意, 要使用/作为路径分格符.
如果你想在任意目录下都可以使用这些工具, 可以在用户变量中新建Path变量(如果存在就选中编辑它), 将其值设为%Path%; C:\amber14\bin(如果已存在Path变量, 只要增加C:\amber14\bin即可).
如果你要使用Python版本的ACPYPE, 而不是(或不能)使用我编译好的二进制版本, 到Python官方网站下载Python 2.x安装即可.
对电荷的处理, 目前有两种比较合理的方法, AM1-bcc电荷与RESP电荷. AM1-bcc电荷可以使用AmberTools中的antechamber程序直接得到(或使用Chimera软件, 它集成了antechamber). RESP电荷则需借助第三方量子化学程序来得到, 如Gaussian, GAMESS等. 因此, 如果你想使用RESP电荷, 那你还需要安装一种量子化学程序. 由于Gaussian使用方便, 所以使用更广泛些. Windows版本的Gaussian及其自带的GaussView界面安装很容易, 网上资料也很多, 这里不做介绍.
使用AmberTools+ACPYPE+Gaussian创建小分子GAFF力场拓扑文件的整个流程如下:

创建过程的大致步骤是先利用Gaussian得到RESP电荷, 然后利用AmberTools得到AMBER的参数文件. 由于GROMACS和AMBER参数文件的格式很不一样,所以最后需要使用ACPYPE将AMBER参数文件转换为GROMACS可识别的.gro和.top文件. 具体步骤说明如下:
1. 使用GaussView创建分子构型并做初步优化
使用熟悉的分子编辑软件创建分子构型. 可用的软件非常多, 一般只要支持输出为.pdb或.mol2格式即可. GaussView, Chimera, Chem3D, VMD等等都可以. 由于我们需要使用Gaussian计算静电势以用于拟合RESP电荷, 所以使用与Gaussian配套的GaussView来构建分子并准备输入文件更方便些. 因此, 如果你对分子编辑软件还没有形成什么偏好的话, 那我推荐你试一试GaussView.
我这里使用的Gaussian 09 C.01版本, 建议你至少使用这个版本, 或使用更新的D.01版, 不要使用更低的版本, 因为下面的有些操作更低的版本不支持. 详细信息见参考资料中的博文.
构建好分子之后, 一般要做下粗略的优化, 使搭建出来的分子构型看起来更合理一些. 这在GaussView中很容易完成, 只要点击Edit -> Clean就可以了.
注意, Gaussian以及GaussView的安装路径中不能含有中文, 输入输出文件的保存路径中也不能使用中文
下面就是我们构建出来的分子结构
2. 使用Gaussian进一步优化分子构型并计算静电势
粗略优化之后, 我们要使用Gaussian进行进一步的优化, 同时计算其静电势, 用于后面拟合RESP电荷. 如果你熟悉Gaussian的计算流程, 可以先保存文件, 然后修改输入文件后在命令行中运行. 如果不熟悉的话, 你可以在GaussView的菜单中操作.
点击Calculate->Gaussian Calculation Setup, 打开输入文件编译界面
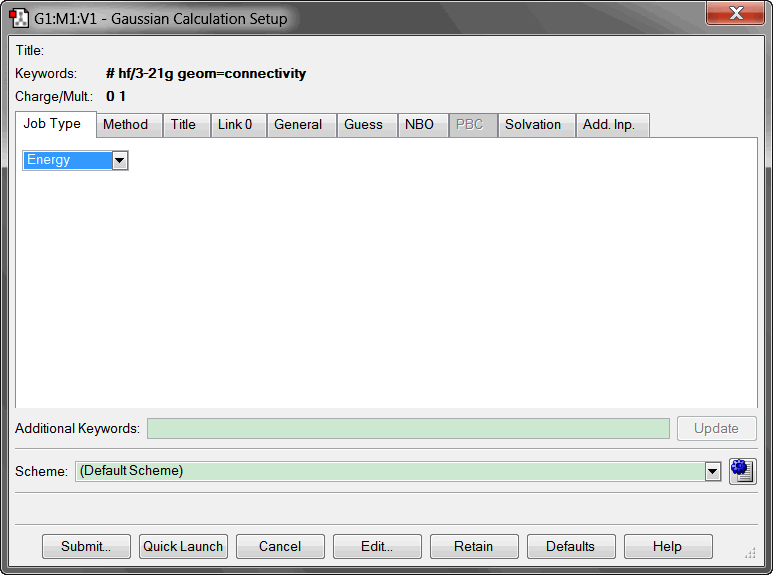
先将计算类型修改为优化,
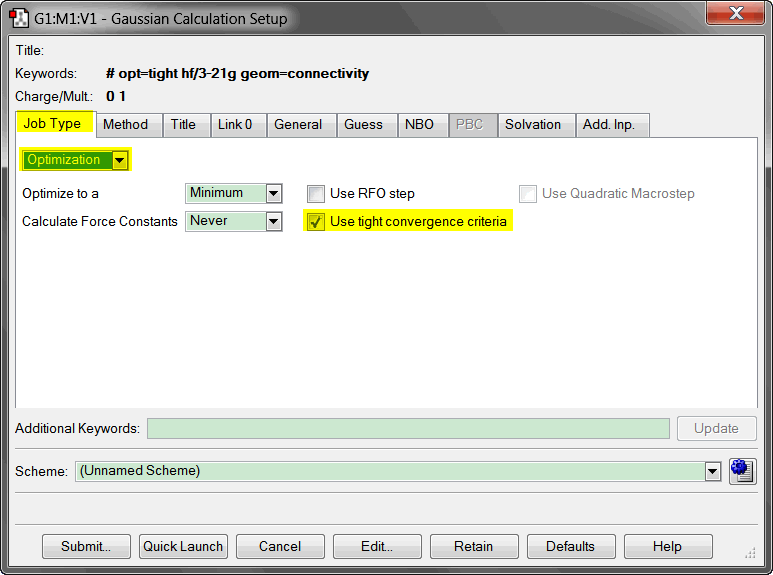
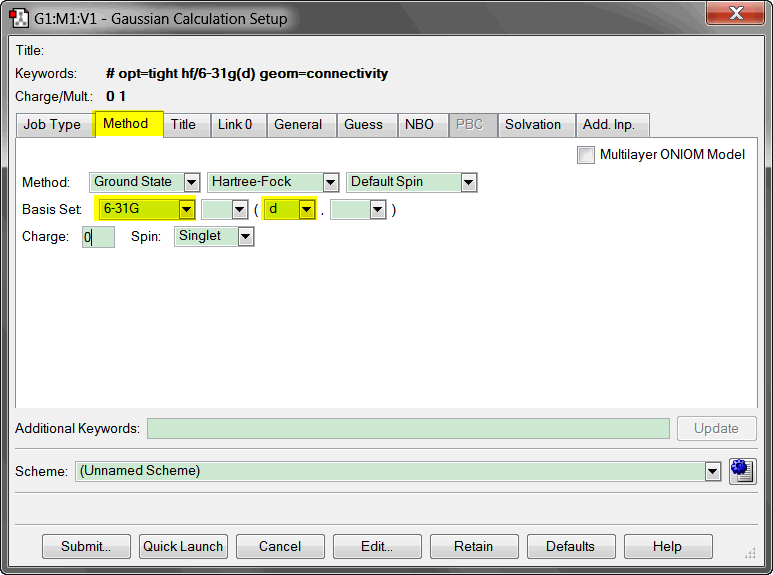
设定优化使用的方法, 基组, 以及体系的电荷, 自旋多重度
标题段可改可不改
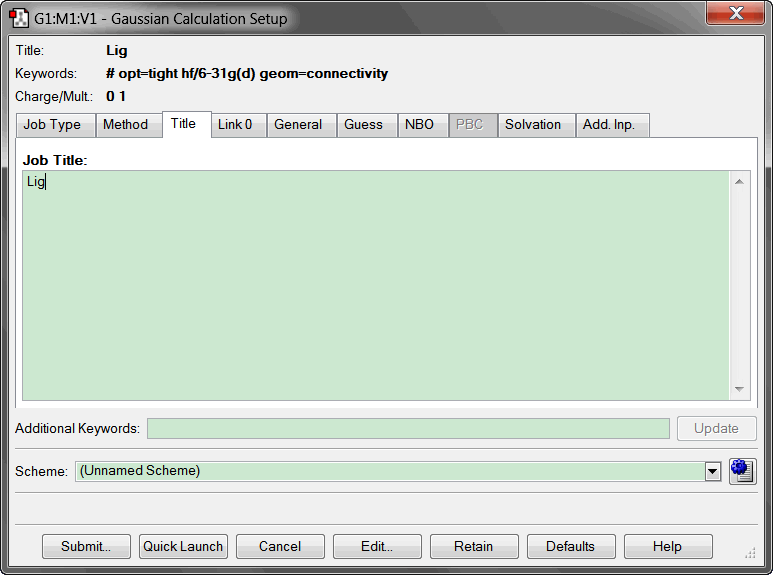
Link 0部分可设定计算时所用的内存和核数. Windows下Gaussian最多可使用1 GB内存, 我的电脑是四核的, 这里我设置使用2个核
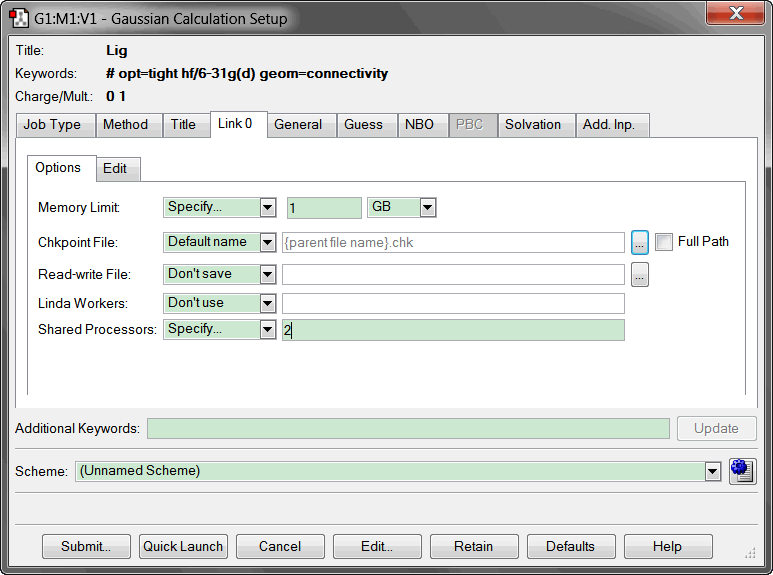
General部分设置使用二次收敛的SCF方法以确保收敛, 选择忽略对称性, 不将连接信息写到输入文件中. 其实这些设置影响不大, 不改一般也没事.
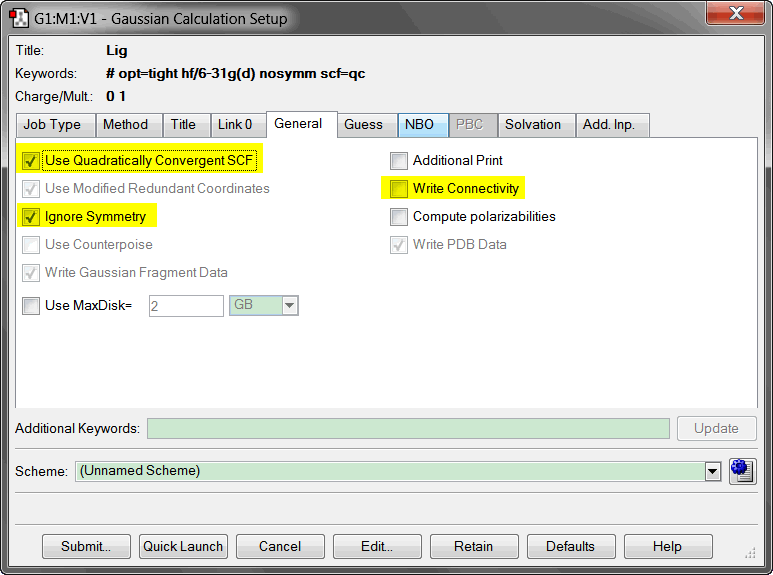
最后, Add. Inp.部分添加静电势输出文件的名称, 并在``中添加计算静电势的关键词
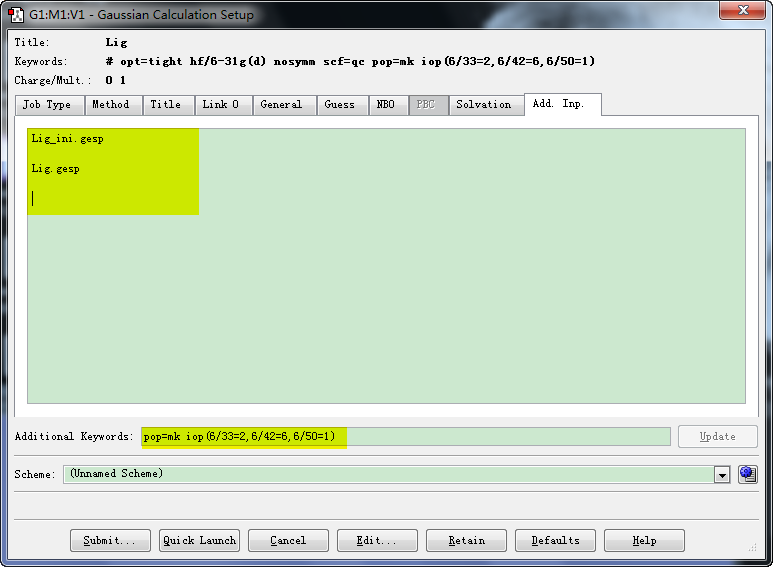
点击Submit..保存为.gjf文件, 并输出直角坐标和附加输入
打开产生的输入文件, 内容如下:
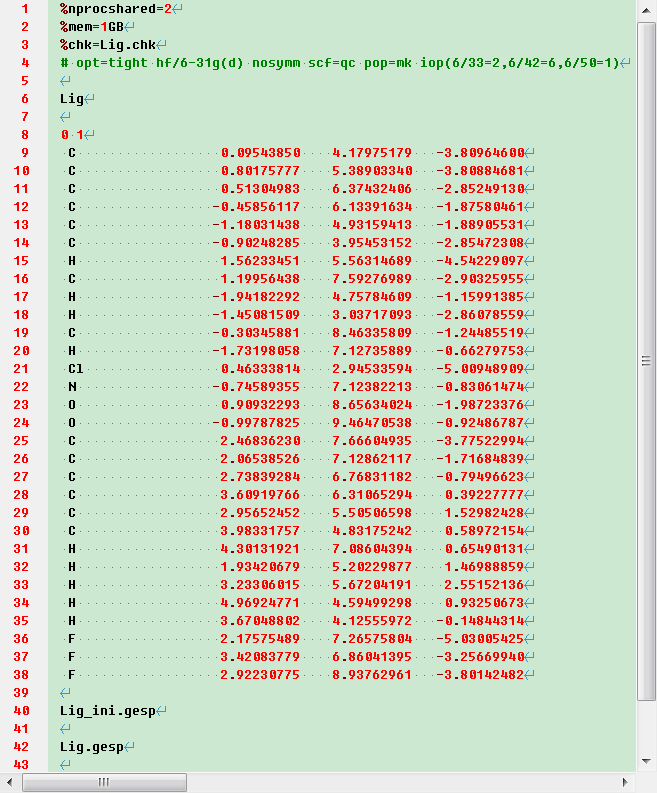
有关输入文件中关键词的解释, 见参考资料中的博文.
注意, Gaussian输入文件中的空行非常重要, 要严格按图上的格式来. 比如, Lig_ini.gesp前面只能有一个空行, 而Lig.gesp后面至少要有两个空行.
得到了输入文件后, 我们就可以使用Gaussian来运行它了. 你可以使用Gaussian自带的用户界面来运行, 也可以使用命令行.
由于这个分子较大, 在我的机器上, 运行了3个多小时才正常结束.
运行正常结束后, 会产生Lig.chk, Lig.out, Lig_ini.geps和Lig.gesp等文件, 我们只需要其中的Lig.gesp文件, 它是优化后构型的静电势文件, 我们就使用它来拟合RESP电荷.
另外, 我们也可以利用antechamber命令来产生运行Gaussian需要的输入文件, 像下面这样
antechamber -i Lig.mol2 -fi mol2 -o Lig.gjf -fo gcrt -pf y -gm "%mem=1GB" -gn "%nproc=2" -nc 1 -gk "#HF/6-31G* SCF=tight Pop=MK iop(6/33=2,6/42=6,6/50=1)"
3. 使用antechamebr拟合RESP电荷
使用上一步得到的Lig.gesp文件, 运行antechamebr拟合RESP电荷
antechamber -i Lig.gesp -fi gesp -o Lig.mol2 -fo mol2 -pf y -c resp
我们只需要Lig.mol2输出文件, 它包含了构型以及RESP电荷, 其他文件ANTECHAMBER*, ATOMTYPE.INF BCCTYPE.INF NEWPDB.PDB PREP.INF, esout, qout, punch等都可删除.
4. 使用parmchk2检查GAFF参数并生成缺失参数文件
使用上一步得到的Lig.mol2文件, 运行parmchk2命令
parmchk2 -i Lig.mol2 -f mol2 -o Lig.frcmod
parmchk2是原先parmchk的增强版, 可以检查输入分子构型中GAFF的缺失参数, 并生成相应的补充参数文件Lig.frcmod.
5. 使用sleap生成AMBER参数文件及坐标文件
编写一个leap.in文本文件, 内容如下:
| bash | |
|---|---|
1 2 3 4 5 6 7 | source leaprc.ff14SB
source leaprc.gaff
loadamberparams Lig.frcmod
lig=loadmol2 Lig.mol2
check lig
saveamberparm lig Lig.prmtop Lig.inpcrd
quit
|
然后运行sleap命令
sleap -f leap.in
这样就拿到了分子的AMBER参数文件Lig.prmtop, 结构文件Lig.inpcrd.
也可直接打开sleap依次执行leap.in文件中的每一行, 只不过麻烦一些.
6. 使用ACPYPE将AMBER文件转换为GROMACS文件
运行ACPYPE命令
acpype -p Lig.prmtop -x Lig.inpcrd -d
这样就得到了GROMACS支持的Lig_GMX.gro, Lig_GMX.top, em.mdp, md.mdp等文件. 一般我们只需要前面两个文件.
如果想将.top文件进行处理生成.itp文件,以便在蛋白质的拓扑文件中包含, 可以除去表头, 改动原子类型, 再除去后面的附加信息.
实际上, 上面的3, 4, 5, 6这几个步骤可以使用ACPYPE一步完成, 但在Windows下由于路径的原因很容易出问题. 像上面这样分开做的话, 一步一步完成的话, 出错了容易定位具体的出错步骤. 如果你对这个过程很熟悉了, 可以将这些流程写在一个脚本中自动执行, 或是研究一下如何使用acpype一步执行成功.
【2016-03-28 补注】
Google上搜索acpype code, 主要有两个版本供下载: GitHub版和SF版. 前者是个旧版本, 与后者的主要区别在于生成的拓扑文件中二面角的默认函数类型. 旧版本生成的拓扑文件中, 二面角的默认函数类型为3或1, 但可通过-r选项改变. 而新版本中, 二面角的默认函数类型为9或4, 可通过-z选项改变. 由于GAFF力场中二面角的函数类型为9或4, (文档说明), 所以建议使用新版本的ACPYPE. 我提供的压缩包中包含的是新版本. 有关新版本的说明可参考这里.
参考资料
- AMBER:小分子处理
- GMX中如何实现小分子的GAFF立场
- 自动计算ESP和RESP电荷(AMBER and G09)
评论
- 2016-02-24 11:44:31
zzz你好,请问如何在高斯中生成gesp文件呢,文章中的命令不能生成啊… - 2016-02-24 23:26:03
Jerkwin仔细阅读, 好好思考. - 2016-02-25 23:09:53
zzz我用的是高斯B版本,输入文件中空行按照文章中的来,可是最后没有得到那两个gesp文件…其它部分都是按照文章中的完全一样 - 2016-02-25 23:57:31
Jerkwin文章中说得很明白: 我这里使用的Gaussian 09 C.01版本, 建议你至少使用这个版本, 或使用更新的D.01版, 不要使用更低的版本, 因为下面的有些操作更低的版本不支持. 详细信息见参考资料中的博文. - 2016-02-26 17:03:42
zzz哦哦,谢谢啦! - 2016-02-26 17:06:43
zzz接楼主的帖子提醒下大家另一件事:计算ESP电荷不要使用高斯的B版本,这个版本计算ESP电荷有bug,使用G09 a c d都可以,G03也可以。 -
2016-02-26 22:58:57
Jerkwin参考资料中的博文说明了低版本的为什么不行. 我自己没有试过, 不确定你的说法. 你要是对比过这些版本, 发现得到的结果相同, 请告诉我, 这样我可以更新文章. - 2016-07-11 15:14:43
切嗣耙耙_少年哟成为神话吧老师您好,能否解释一下这一段话里 默认函数类型是什么意思? 谢谢 “旧版本生成的拓扑文件中, 二面角的默认函数类型为3或1, 但可通过-r选项改变. 而新版本中, 二面角的默认函数类型为9或4, 可通过-z选项改变. ” -
2016-07-14 14:23:06
Jerkwin这个你要去看gmx手册的第四章二面角的函数类型那部分, 如果你没有了解过力场的实现, 很难说明白 - 2016-07-28 16:37:50
卡尔啾啾老师,为什么用amber14文件夹里的ACPYPE命令会闪退啊,里面的好多软件都会闪退,我试过winxp32、win7 64 、win10 64都试过 -
2016-07-28 17:06:37
Jerkwin你说的闪退是不是指双击后直接闪退? 如果是的话, 那说明你要在CMD下运行才可以. 否则的话, 要看具体的出错信息. 这个工具我在XP32和win7 64下测试过, 没有问题. -
2016-08-22 16:50:39
tianflame李老师您好,我使用您编译好的amber14,利用antechamber命令对一个小分子ack.pdb产生prepi文件,指定bcc电荷,结果报错,输入命令如下,您能帮忙看一下是什么原因吗?谢谢! D:\amber14\bin>antechamber -i ack.pdb -fi pdb -o ack.prepi -fo prepi -c bcc -at amber Total number of electrons: 102; net charge: 0Running: D:/amber14/bin/sqm -O -i sqm.in -o sqm.out sh: 1: cp: not found Error: cannot run \"cp -rf ANTECHAMBER_PREP.AC0 ANTECHAMBER_PREP.AC\" in wprep() of prep.c properly, exit - 2016-08-23 02:37:46
Jerkwin这个工具我只用来做resp电荷, 不是用来替代amber的. 真要做amber的话, 建议你安装完整版的amber. 具体到你的问题, 是因为你的windows系统下没有安装bash环境造成的. 你安装一个bash环境, 然后将其路径添加到path环境变量就可以了. bash环境可以用msys2或cygwin. -
2016-08-25 10:10:05
tianflame好的,非常感谢! - 2016-10-25 08:00:17
LR我想问一下,可以在linux系统下安装吗 - 2016-10-25 15:12:28
Jerkwin所用的工具linux下都有, 安装差不多, 使用几乎完全一样, 没有难度. -
2016-10-25 21:30:44
LR好的,谢谢 - 2017-02-14 15:11:27
.老师您好,你在第6步中说的“可以除去表头, 改动原子类型”,其中,改动原子类型是什么意思? - 2017-02-14 15:17:10
.哦,改动原子类型就是将“atomtypes”放入总top文件里面,而itp文件中是从“moleculetype”开始的,对吗? -
2017-02-14 19:58:37
Jerkwin大致是你说的意思, 但根据实际情况可能有不同的作法. 你清楚拓扑文件的结构就明白该怎么做了. - 2017-03-10 18:39:19
tianflame李老师你好,请问在第3步得到Lig.mol2文件后,是否可以直接用acpype处理Lig.mol2得到gromacs所需的top,gro和itp文件? -
2017-03-11 01:22:04
Jerkwin应该可以, 因为acpype可以一步完成的. 理论上说, 从任何一步开始, 都能直接得到最终的拓扑文件. 但具体如何实现, 需要测试. - 2017-04-06 19:20:45
高庆林g09Wa生成不了gesp已验证,换d版Linux - 2017-04-06 20:04:37
Jerkwin谢谢告知. - 2017-04-07 12:42:53
高庆林老师A01我忘了加iop了,但是现在验证了Gaussian 09: ES64L-G09RevD.01可用 -
2017-04-07 22:02:45
Jerkwin好, 有D版本的话, 建议用D版本. - 2017-04-11 09:48:48
高庆林老师,生成了单个离子液体对的pro,和top文件,但是想生成多个离子液体对的top文件该如何进行,我刚开始想把这个分子变成itp文件包含进去,但是坐标文件,和top文件该如何更新呢?已经困在这里好几天了 -
2017-04-11 19:49:20
Jerkwin同类型的分子itp是相同的, 总top中#include想要的itp, 写明分子个数, 至于gro文件, 可以用gmx solvate或packmol来做. - 2017-04-21 21:30:06
Lewis老师您好!我在运行antechamber -i -fi gesp -o -fo mol2 -pf y -c resp后提示说 cannot open a fileto write in ac format, exit 不知是什么意思? - 2017-04-22 12:14:26
Jerkwin-i 后面指定输入文件啊, 仔细阅读教程. - 2017-04-23 16:27:09
lewis抱歉老师我没说清楚,我是按照文章中的规范输入的,这里只是简写。我在linux中运行这一步成功了,可能是我在win下面没有安装好吧 -
2017-04-24 11:08:00
Jerkwin环境变量没设置好 - 2017-04-21 15:18:29
二马老师您好,打扰您了。我按照您的方法生成我的 XXX .gesp 文件之后,在antechamber 里面输入 antechamber -i XXX.gesp -fi gesp -o Lig.mol2 -fo mol2 -pf y -c resp 之后出现了报错,如下所示,想请问下老师,我这是出现了什么问题,是不是antechamber 没安装好? 谢谢! ************************* Warning: $AMBERHOME and $ACHOME enviornment strings are not set, use "mopac.sh" in the work directory Cannot find atom type defination file!, define with "-def" option or set ACHOME or AMBERHOME environment Error: cannot run "atomtype -i ANTECHAMBER_AC.AC0 -o ANTECHAMBER_AC.AC -p gaff" in main() of antechamber.c properly, exit -
2017-04-21 20:01:21
Jerkwin环境变量没设好, 对照前面部分把AMBERHOME环境变量设好 - 2017-04-22 16:24:32
二马感谢老师您百忙之中的回复,我之前是在linux上自己摸索的。现在按照您的文章,成功安装了amber14,环境变量也仔细检查了,可是运行时会出现如下的报错,还想跟老师请教下原因,谢谢! ********* sh: 1: rm: not found Error: cannot run "rm -f ANTECHAMBER* ATOMTYPE.INF BCCTYPE.INF NEWPDB.PDB PREP.I NF" in main() of antechamber.c properly, exit - 2017-04-22 22:05:57
二马老师您好,我跟您一样安装在了C盘目录下,我仔细检查了下,(%Path%; C:\amber14\bin )这个也设置好了。我发现我修改C:/amber14 变成C:\amber14,或者C:/amber,都会出现同样的报错这个设置我也仔细检查了问题,请问老师还是我环境变量设置的问题吗?或者是其他的原因。谢谢! - 2017-04-22 22:16:30
二马我仔细看了下其他的评论,是.bash环境的问题吗?我试了3个Win7的电脑都不可以,我去安装下.bash试一试 -
2017-04-24 11:07:00
Jerkwin仔细看教程, 只要得到了正常的mol2文件就可以了, 其他中间文件不能自动删除的话, 可手动删除. 这个报错是因为你没有bash环境, 无法执行rm命令, 不影响得到mol2文件 - 2017-05-03 21:07:08
二马谢谢老师您百忙之中的指点,学生安装了bash环境之后解决了问题。还想跟您请教下,学生在win下用G09D 计算时会出现2070 错误,报错如下,网上说用Linux算就不会出错,是这样吗?谢谢! ***************** No lower point found – switch to steepest descent. Search did not lower the energy significantly. Search did not lower the energy significantly. Search did not lower the energy significantly. No lower point found – run aborted. Error termination via Lnk1e in D:\Program Files (x86)\Gaussian\G09W\l508.exe at Wed May 03 10:53:53 2017. Job cpu time: 4 days 13 hours 5 minutes 34.0 seconds. - 2017-05-04 22:24:48
Jerkwin看起来你这是优化出错, 抽取最后的构型看看优化成什么样子了. 如果还算正常的话, 继续优化就是了, 实在优化不出来, 再找其他原因. -
2017-05-10 21:12:42
二马嗯,感谢老师的回复! - 2017-05-11 15:01:55
二马老师您好,打扰您了。我查到一篇参考文献是这么说的“HF/6-31G* RESP charge”,它模拟了醇类的gaff 力场下的 RESP电荷,我根据您的文章,在gaussian 里面导入一个分子的pdb,设置跟您一样,就是一个地方改成了 HF/6-31G* 。但是算下来的结果,比如 oh ,文献中是-0.6e,我算下来有-0.8e,如果老师方便的话,想跟老师您请教下原因。谢谢!
