- 2016-05-31 11:22:44 整理: 刘世恩; 补充: 李继存
- 2016-06-01 10:37:24 补充VMD的pbc命令: 赵道辉
使用GROMACS完成模拟后, 有时需要对轨迹的周期性边界条件(PBC)进行处理. 可能的目的主要有两个: 为了观看轨迹时分子保持完整, 不产生断键或过长的键, 同时未解离的复合物保持在一起; 为了对选定的中心分子进行居中, 以便进行后面的分析. GROMACS的大多数分析工具都会自动处理PBC, 不劳自己费心, 如计算距离, 角度, RDF, MSD时, PBC会自动考虑在内, 所以没有必要对轨迹进行预处理. 但对那些分析时需要进行叠合的工具, 如计算RMSD之类, 需要对轨迹进行预处理, 这种情况下就需要小心处理PBC的问题.
GROMACS处理PBC的主要工具是gmx trjconv, 请先仔细阅读其文档及补充说明, 确保理解-pbc几个选项的功能, 以及-center的作用. 如果不能明白其中的道理, 在处理轨迹时只是尝试选项的各种组合, 一味地乱试, 很难得到正确的结果.
下面我们以双链DNA体系为例, 具体说明一下如何使用gmx trjconv来处理复合物体系. 这里所谓的复合物, 指的是体系中有一些分子在模拟过程中会聚集在一起, 并不解离. 比如, 模拟双链DNA时, 如果DNA的两条链始终没有分开(一般是这种情形, 否则的话, 所用的力场可能有问题), 就可以认为它们形成了一个复合物. 我们在观看轨迹时, 不希望看到DNA的两条链在盒子中分离开来, 这时就需要进行PBC的处理了. 模拟蛋白-配体复合物, 或者分子聚集体如胶束时, 有时也会面临的同样的情况.
首先需要明确, 只要在整个模拟过程中复合物没有解离, 我们一定可以通过PBC处理使其处于盒子中间且保持完整; 如果模拟过程中复合物发生了解离, 但解离分子之间的最大距离小于盒子长度的一半, 我们仍可以通过PBC处理使这些解离的分子处于盒子中, 不在盒子两侧跳来跳去; 如果解离分子之间的最大距离超过了盒子长度的一半, 我们还可以通过PBC处理使每个解离分子保持完整, 但没有办法保证观看轨迹时所有的分子不在盒子两侧之间跳跃. 其中的原因, 想想PBC的定义就能明白.
具体的示例体系是一段双链DNA, 并添加了离子和水(为清晰起见下面的图中忽略了水和离子), DNA中的原子距盒子边界的最小距离为1 nm.
使分子保持完整
模拟完成得到轨迹后, 按照经典的轨迹处理步骤, 先保证体系中每个分子完整:
gmx trjconv -s npt.tpr -f prod.xtc -o prod_whole.xtc -pbc whole
使用VMD观看得到的轨迹prod_whole.xtc, 则发现虽然每个分子都保持了完整, 没有异常的键, 但对很多帧, DNA两条链不在盒子内, 而是分处于盒子两侧, 且X, Y, Z三个方向均出现这种情况. 在播放轨迹时两条链不时地在盒子两侧之间跳跃. 根据经验, 初步认为这是由于PBC处理不当导致的.
为确认这一点, 我们使用VMD查看轨迹中的一帧test.pdb. 在VMD的命令窗口中执行pbc box显示盒子, 可看到两条链沿X轴方向确实分处于盒子两侧, 且有些原子处于盒子内, 有些处于盒子外.
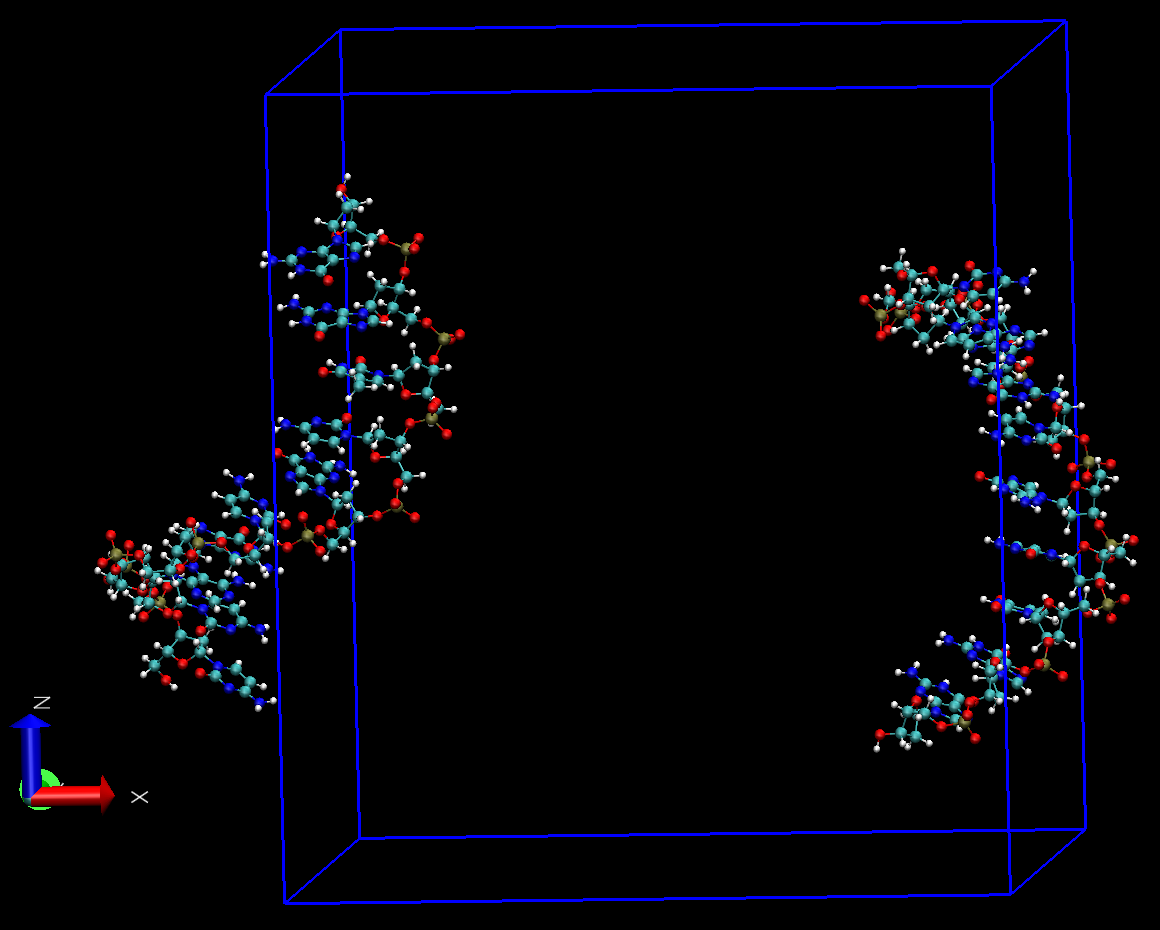
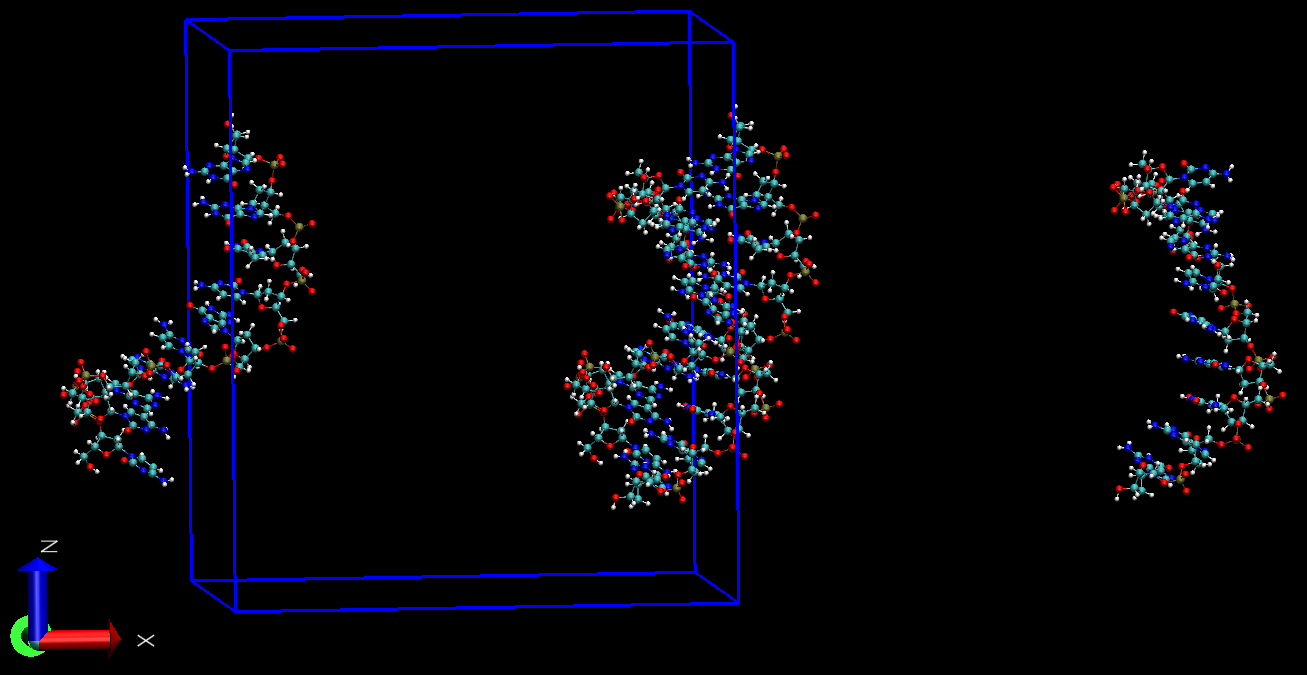
为确认两段DNA并没有分离, 点击Graphics -> Representations -> Periodic, 选择+X或-X体系在X反向的映象, 就可看到两条链处于一起了, 这就说明先前显示的分离确实是由于PBC的原因导致的. 根据上面的说明, 我们一定有办法通过PBC处理使VMD显示正常.
使复合物按中心原子进行居中
对得到的轨迹prod_whole.xtc进行下一步处理时, 需要选择DNA分子中需要居中的 一个原子. 选择的标准是, 如果将DNA以此原子在盒子中居中, DNA中的所有原子均能包含在盒子中. 这里要注意的是, 对我们的这个体系, 不要选择序列最中间配对的两个核苷酸(或其C1’)作为中心组, 因为这样的话, 会以这些原子的质心进行居中, 而这些原子已经分离开来, 即便以其质心居中, 仍不能保证所有原子都聚集在一起.
我们选择154号原子作为中心, 因为它近似处于分子的中心
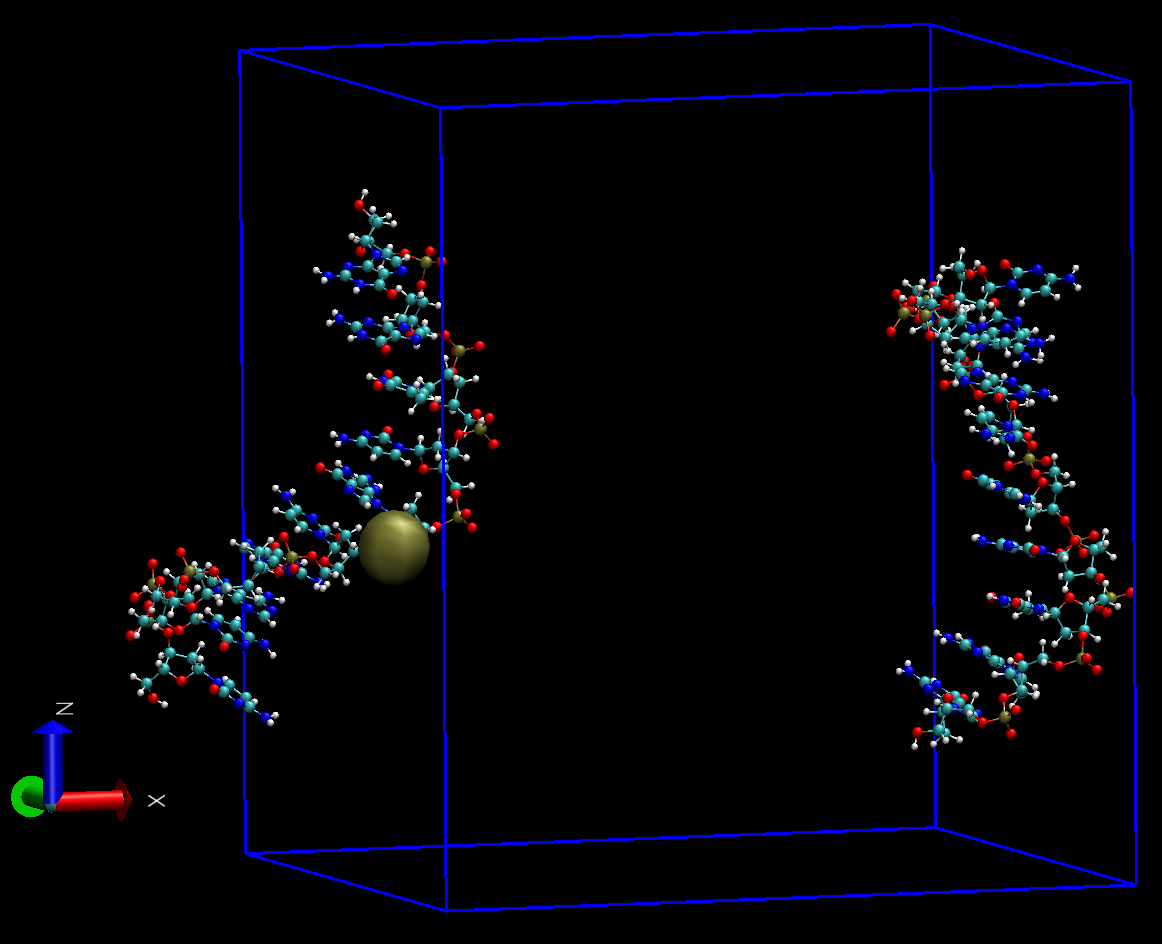
在index.ndx文件中添加这个中心组
[ center ]
154
然后执行下面的命令
gmx trjconv -s npt.tpr -f prod_whole.xtc -n index.ndx -o prod_atom_center.xtc -pbc atom -center
提示时分别选择中心组和DNA即可. test.pdb处理后的构型如下
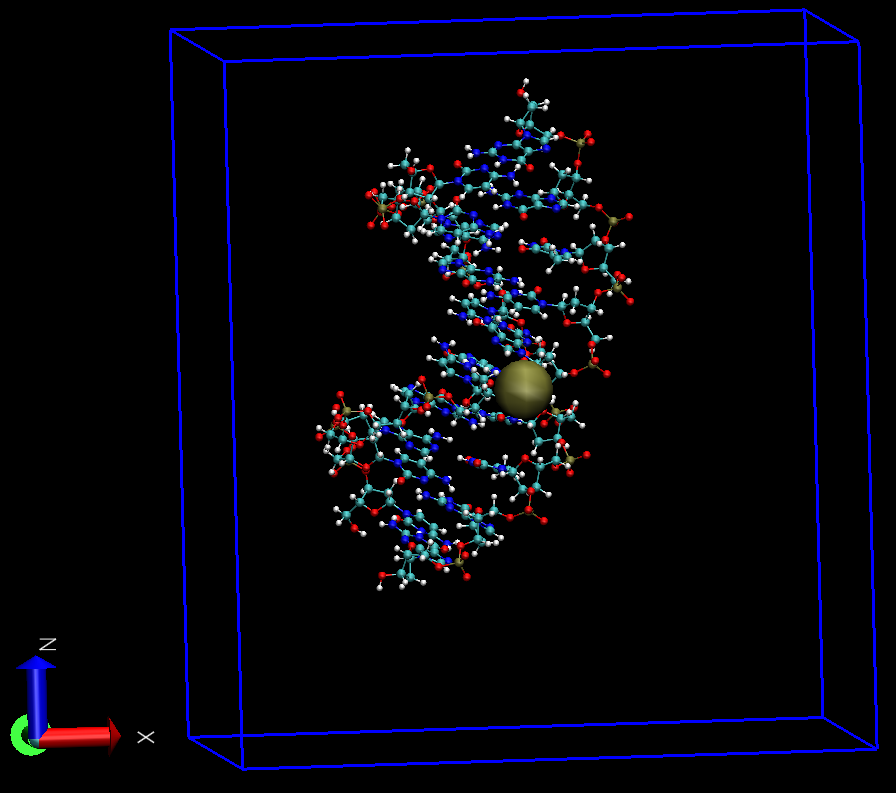
关于上面命令中-pbc选项的选取, 则根据不同情况来,
-
atom: 最通用, 适用于所有情况. 但如果查看得到的轨迹, 发现个别帧仍有部分原子处于盒子外面, 则说明所选的中心原子不合适, 需要重新选择中心原子.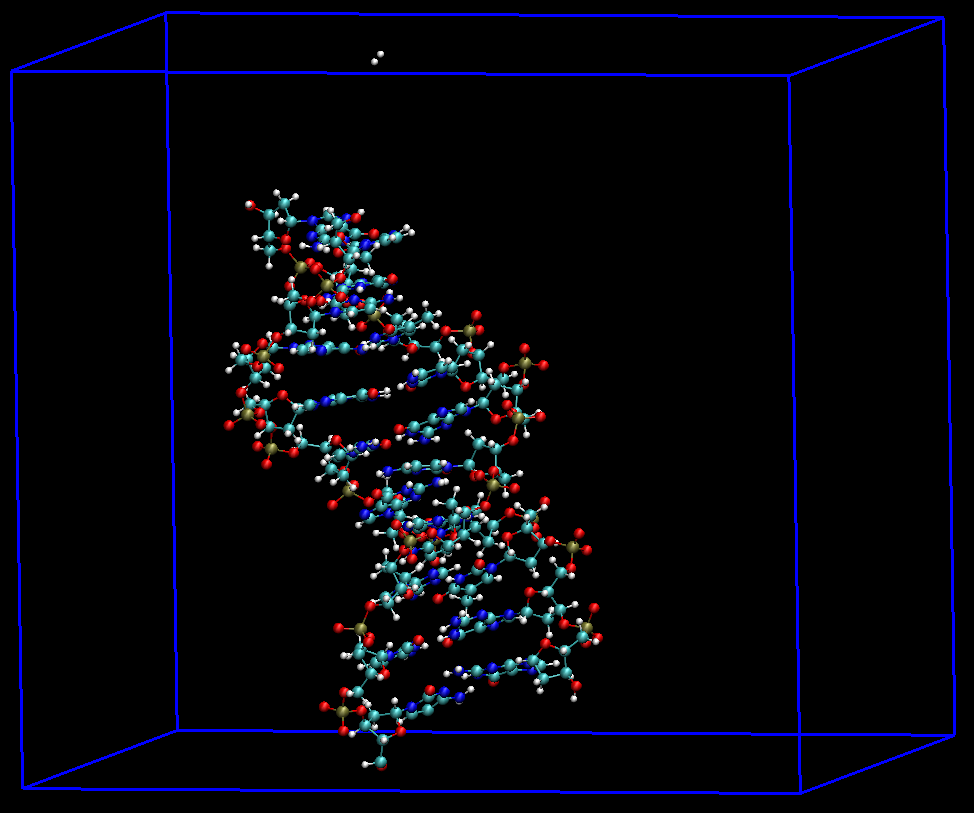
此外, 若复合物距离盒子边缘的距离过小, 也容易出现这种情况, 因此建议准备体系时, 盒子尽量大些, 视个人计算资源而定, 一般可取3-5 nm. 一则处理PBC时方便, 二则可以避免计算能量时引入分子映象间相互作用的误差.
res: 用于生物分子, 因为在这些分子中可以定义残基. 对我们的示例DNA体系, 使用此选项可能比atom更好.mol: 只有当复合物中的每个分子在拓扑文件的[ system ]中单独定义时, 使用这个选项才有效. 对我们的DNA体系, 由于在拓扑文件中将两条链定义为了一个分子类型,gmx trjconv处理时会将两条链视为同一个分子, 即便分子质心处于盒子中, 其中某条链或者两条链仍可能处于盒子外, 所以使用此选项不能满足要求.
如果需要, 进行叠合处理
如果需要, 可以进一步进行叠合去除平动和转动
gmx trjconv -s npt.tpr -f prod_atom_center.xtc -o prod_atom_center_fit.xtc -fit rot+trans
附
如果你想要尝试一下上面作法, 可以下载用到的文件.
另: 使用VMD的pbc命令显示完整分子
VMD提供了一个pbc命令, 也可用于对体系的PBC进行处理. 如果只是用于论文作图, 使用这个命令可能比上面的方法更简单.
基本命令是
pbc wrap -compound res -all
pbc box
你也可以同时对盒子进行平移, 以将分子显示在盒子中央(注意, 平移是以盒子长度为单位的)
pbc wrap -compound res -all -shiftcenterrel {0.0 -0.5 0.0}
pbc box -shiftcenterrel {0.0 -0.5 0.0}
如果溶质分子的原子类型都是1, 你可以使用下面的命令使其在盒子中居中
pbc wrap -sel type=1 -all -centersel type=2 -center com
更复杂的一个命令
pbc wrap -center com -centersel protein -compound fragment -all
详细说明请参考其文档
评论
- 2016-11-07 19:26:04
franklinly太好了,李老师,只是这里面“如果你想要尝试一下上面作法, 可以下载用到的文件.”,下载后发现没有prod.xtc文件。试不了啊,希望李老师补上,谢谢。franklinly - 2016-11-08 08:25:12
Jerkwin有了输入文件, mdrun一下就可以了. 轨迹文件太大, 没法放附件.
