- 2016-08-05 15:40:42
Packmol是一个常用的用于构建复杂体系的程序, 使用简单, 速度不错. 这里是翻译并整理的其官方网站上的一些资料, 供参考.
点击这里下载我编译好的Packmol程序(Windows 7 64位)以及自带的示例文件.
- PACKMOL: 利用堆积优化方法创建分子动力学模拟所需的初始构型
- 用户手册
- 使用示例
- 网络资料
- 评论
PACKMOL: 利用堆积优化方法创建分子动力学模拟所需的初始构型
PACKMOL 通过在定义的空间区域中堆积分子, 为分子动力学模拟创建所需的初始构型. 同时, 程序会对分子的堆积方式进行优化, 以保证分子间的短程排斥相互作用尽可能小, 这样进行模拟时体系不至于炸散.
程序支持各种可用于分子或分子中原子的空间约束方法, 因此使用它可以很容易地构建有序体系, 如层状, 球形或管状脂质层.
使用时, 用户只需要提供每种分子类型的一个分子坐标, 每种类型分子的数目, 以及每种分子类型满足的空间约束条件即可.
程序兼容PDB, TINKER, XYZ和MOLDY格式的输入文件.
用户手册
注意: 请下载使用最新版本的Packmol以保证可以使用程序所有的功能.
1. 准备工作
你需要准备好模拟需要的每种分子类型的坐标文件. 例如, 如果你要模拟水和离子的溶液, 那你就要准备 单个 水分子的坐标文件, 以及每种离子的坐标文件. 不同分子类型的坐标文件是独立的, 支持的格式有PDB, TINKER, MOLDEN或MOLDY.
当然, 你也需要Packmol程序, 你可以到http://www.ime.unicamp.br/~martinez/packmol网站, 点击下载链接, 下载源代码packmol.tar.gz .
如果你打算使用MOLDY进行模拟, 请参阅此文档.
2. 如何编译Packmol
从Packmol主页下载packmol.tar.gz压缩包后, 你需要先解压文件, 然后进行编译. 根据你所使用的操作系统, 步骤有所不同.
Linux系统
使用下面的命令解压文件
tar -xvzf packmol.tar.gz
会得到目录packmol, 里面包含源代码.
使用下面的命令构建可执行程序
cd packmol
make
如果没有发生错误, 就得到了可执行文件.
Windows系统
如果你使用CygWin进行编译, 步骤与Linux相同.
如果你使用Windows下的Fortran编译器(如ifort)进行编译, 可按Makefile中的文件顺序依次进行编译, 最后再链接为可执行程序. 或不断执行ifort -c *.f *.f90 *.obj, 至每个源文件都生成.obj目标文件后, 再ifort -o packmol *.obj. 也可以使用我修改的批处理脚本
| batch | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 | @ echo off
set FORTRAN="C:/Program Files (x86)/Intel/Composer XE 2015/bin/intel64/ifort.exe"
set FLAGS="-O3 -ffast-math"
set src= ^
sizes.f90 ^
compute_data.f90 ^
input.f90 ^
flashmod.f90 ^
usegencan.f90 ^
swaptypemod.f90 ^
ahestetic.f90 ^
cenmass.f90 ^
initial.f90 ^
title.f90 ^
setsizes.f90 ^
getinp.f90 ^
strlength.f90 ^
output.f90 ^
checkpoint.f90 ^
writesuccess.f90 ^
fparc.f90 ^
gparc.f90 ^
gwalls.f90 ^
comprest.f90 ^
comparegrad.f90 ^
packmol.f90 ^
polartocart.f90 ^
resetboxes.f90 ^
tobar.f90 ^
setibox.f90 ^
restmol.f90 ^
swaptype.f90 ^
heuristics.f90 ^
flashsort.f90 ^
jacobi.f90 ^
pgencan.f90 ^
gencan.f ^
random.f90 ^
computef.f90 ^
computeg.f90
for %%i in (%src%) do (
echo %%i
%FORTRAN% %FLAGS% -c %%i
)
%FORTRAN% %FLAGS% -o packmol *.obj
del /F /Q *.mod *.obj
pause
|
在Linux下进行*编译时, 如果出现问题, 首先使用configure脚本查看合适的编译器
chmod +x ./configure (使脚本可执行)
./configure (执行脚本)
如果使用此脚本仍无法找到合适的编译器, 你可以手动指定编译器
./configure /编译器/安装/路径/可执行程序
然后执行make. 如果没有错误, 就会得到可执行程序packmol.
运行Packmol
一旦编译成功得到了可执行程序packmol, 可将其路径添加到path环境变量以方便使用. 然后创建输入文件, 使用下面的方法运行Packmol
packmol < packmol.inp
其中packmol.inp为输入文件(你可以到此链接下载示例文件).
如果运行成功, 程序结束后会在屏幕上输出类似下面的文字
------------------------------
Success!
Final objective function value: .22503E-01
Maximum violation of target distance: 0.000000
Maximum violation of the constraints: .78985E-02
------------------------------
其中目标距离的最大异常值(maximum violation of the target distance)表示用户设定的原子间的最小距离, 与程序优化得到构型中原子间最小距离这两者之间的差值. 这个值不会大于10-2. 约束的最大异常值(maximum violation of the constrains)也不能超过10-2.
一个比较好的做法是, 在输入文件中使用check关键词来检查约束是否正确. 使用这个选项, 程序会创建所需结构的一个粗略的初始近似, 但不会进行实际的堆积. 你可以查看输出, 检查分子是否在需要的区域内(但这时不要期望结构很好!). 为此, 你只要将关键词check添加到输入文件的任意一行就可以来(自28 Feb 2008版本起支持此功能).
常见问题
-
运行Packmol时, 如果得到的输出是
Command not found(Linux下)或'packmol' 不是内部或外部命令,也不是可运行的程序或批处理文件。(Windows下), 请使用./packmol < packmol.inp运行程序(将./添加到程序前), 或将packmol可执行程序的目录添加path环境变量. -
如果运行后得到的信息是
Killed, 这是因为程序试图申请的用于静态存储的内存超过了实际值. 打开sizes.i文件, 将maxatom参数减小为你体系中实际的原子数目, 重新编译程序, 再试一次.
基本输入结构
最简略的输入文件, 必须包含需要的距离约束容差(对常温常压下的体系, 坐标以埃为单位, 2.0 Å是个不错的值注). 距离约束容差可以通过下面的语句来指定
tolerance 2.0
- 粗粒化模型: 由于模型不是原子化的, 所需的距离容差可能比原子体系通常所用的更大. 但是, 情况并不总是这样. 因为常用的容差可能够大足以保证对体系进行合适的初始堆积. 自15.133版本起, 对不同原子可以使用不同的半径, 这一功能对上述情况可能会有用.
- 环形体系: 一些用户注意到, 使用2.0 Å的距离容差时, 一些环形体系(如甲苯)在堆积过程中可能会互相连接. 如果你发现这种情况, 稍微增加距离容差可能会解决问题. 你也可以试试增加初始的容差, 例如使用
discale 1.5这个额外的关键词. 在这种情况下, 堆积开始时的容差是设定值的1.5倍, 因此可能会避免重叠. 程序运行过程中容差会逐步减少至真实所需值(通常为2.0 Å).
文件也必须包含要创建的输出文件的名称, 通过下面的语句来指定
output test.pdb
还需要指定文件类型(pdb, tinker, xyz或moldy, pdb为默认文件类型),
filetype pdb
文件中至少需要存在一种分子类型, 这可通过structure ... end structure节来设定. 例如, 如果water.pdb是包含单个水分子坐标的文件, 可在你的输入文件中添加类似下面的语句
structure water.pdb
number 2000
inside cube 0. 0. 0. 40.
end structure
这一节指定了2000个分子, 分子类型是water.pdb. 这些分子将被置于一个立方体内, 最小坐标为(x,y,z) = (0,0,0), 最大坐标为(40,40,40).
根据上面的说明, 最简略的输入文件类似下面这样
tolerance 2.0
output test.pdb
filetype pdb
structure water.pdb
number 2000
inside cube 0. 0. 0. 40.
end structure
使用这个输入文件运行Packmol, 就会得到边长为40.0 Å的立方盒子, 其中填充了2000个水分子. 属于不同分子的原子间的最小距离至少是2 Å, 所有分子会随机地分布在立方体内.
多个分子类型
你可以添加多个分子类型到同一区域或空间的不同区域, 只要简单地添加其他structure ... end structure节到输入文件即可.
原子选区
单个分子的坐标文件, 包含例如10个原子. 你可以将分子的一部分限制在空间中的指定区域内. 在构建囊泡时这很有用, 因为其中的表面活性剂的亲水部分必须指向水环境. 对含有10个原子的分子, 可通过使用atom关键词来达到目的,
structure molecule.pdb
inside cube 0. 0. 0. 20.
atoms 9 10
inside box 0. 0. 15. 20. 20. 20.
end atoms
end structure
上面的例子中, 分子的所有原子会被置于定义的立方体中, 但9号和10号原子会被约束在特定的盒子内.
约束类型
有多种约束类型, 它们既可用于整个分子, 也可用于分子的一部分. 这些约束定义了在得到的构型中, 分子必须位于的空间区域. 通过合理使用这些约束可以构建非常有序的结构. Packmol目前支持的约束类型有
1. fixed 固定
用法: fixed x y z a b g
这一选项将分子固定在某一位置, 位置由6个实数参数x, y, z, a, b, g指定, 前三个参数为分子相对于其坐标文件中位置的平移, 后三个参数为分子的旋转角度(以弧度为单位). 对这一选项, 只能设定一个分子. 此约束可能需要与center关键词一起使用. 在这种情况下, 前三个数字表示几何中心的位置(并不是质心, 因为程序假定所有原子的质量都相同). 因此, 这一关键词的必须用于如下的上下文环境
structure molecule.pdb
number 1
center
fixed 0. 0. 0. 0. 0. 0.
end structure
在上面的例子中, 分子的质心将会被固定在原点且不能分子自身不能旋转.
2. inside cube 立方体内
用法: inside cube xmin ymin zmin d
xmin, ymin, zmin和d为4个实数. 所得构型中, 受此选项约束的原子, 其坐标(x,y,z)将满足:
xmin < x < xmin + d
ymin < y < ymin + d
zmin < z < zmin + d
3. outside cube 立方体外
用法: outside cube xmin ymin zmin d
xmin, ymin, zmin和d为4个实数. 所得构型中, 受此选项约束的原子, 其坐标(x,y,z)将满足:
x < xmin 或 x > xmin + d
y < ymin 或 y > ymin + d
z < zmin 或 z > zmin + d
4. inside box 盒子内
用法: inside box xmin ymin zmin xmax ymax zmax
xmin, ymin, zmin, xmax, ymax和zmax为6个实数. 所得构型中, 受此选项约束的原子, 其坐标(x,y,z)将满足:
xmin < x < xmax
ymin < y < ymax
zmin < z < zmax
5. outside box 盒子外
用法: outside box xmin ymin zmin xmax ymax zmax
xmin, ymin, zmin, xmax, ymax和zmax为6个实数. 所得构型中, 受此选项约束的原子, 其坐标(x,y,z)将满足:
x < xmin 或 x > xmax
y < ymin 或 y > ymax
z < zmin 或 z > zmax
6. inside (或outside) sphere 球面内(或外)部
球面由下面的一般方程定义:
\[(x-a)^2+(y-b)^2+(z-c)^2-d^2=0\]因此, 你必须提供四个实数参数, a, b, c和d`来定义球面. 输入语法类似下面这样
inside sphere 2.30 3.40 4.50 8.0
所得构型中, 原子坐标会满足方程
\[(x-2.30)^2+(y-3.40)^2+(z-4.50)^2-8.0^2 \le 0\]其他支持的语法有
outside sphere 2.30 3.40 4.50 8.0
outside的参数类似于inside, 但原子满足的约束方程中使用 $\ge$ 而不是 $\le$, 因此, 原子将位于所定义球面的外部.
7. inside (或outside) ellipsoid 椭球面内(或外)部
椭球面由下面的一般方程定义:
\[{(x-a_1)^2 \over a_2^2}+{(y-b_1)^2 \over b_2^2} +{(z-c_1)^2 \over c_2^2} -d^2=0\]参数类似于球面的例子, 但现在需要7个参数, 且必须按顺序列出
inside ellipsoid a1 b1 c1 a2 b2 c2 d
坐标(a1,b1,c1)定义椭球面的中心, 坐标(a2,b2,c2)定义坐标轴的相对大小, d定义椭球的大小. 同样, 命令
outside ellipsoid a1 b1 c1 a2 b2 c2 d
的使用方式类似于球面. 注意, 当a2=b2=c2=1.0时, 与球面参数一样. 椭球面的参数并没有归一化, 因此, 如果a2, b2和c2很大, 椭球面也或很大, 即便d很小.
8. over (或below) plane 平面上(或下)方
平面由下面的一般方程定义:
\[ax+by+cz-d=0\]可以限制原子处于平面上方或下方, 语法为
over plane 2.5 3.2 1.2 6.2
below plane 2.5 3.2 1.2 6.2
其中, over关键词使原子满足条件
below关键词则使原子满足
9. inside (或outside) cylinder 圆柱内(或外)部
为定义一个圆柱, 首先需要在空间中定义轴线的取向. 在Packmol中, 轴线的定义使用参数方程
\[p = ( a_1, b_1, c_1 ) + t ( a_2, b_2, c_2)\]其中t为独立参数, 向量(a2, b2, c2)定义线的方向. 圆柱由到轴线的距离d和长度l来定义. 使用方法如下
inside cylinder a1 b1 c1 a2 b2 c2 d l
outside cylinder a1 b1 c1 a2 b2 c2 d l
其中, 前三个参数定义了圆柱的起始点, l定义了圆柱的长度, d定义了圆柱的半径. 更简单的一个例子是沿x轴, 起始点为原点的圆柱
inside cylinder 0. 0. 0. 1. 0. 0. 10. 20.
这个圆柱由到x轴距离为10的点定义(圆柱的半径为10), 而且它始于原点. 因此, 受此圆柱约束的原子其x坐标不会小于0. 此外, 圆柱的的长度为20, 因此, 原子的x坐标也不会大于20. 圆柱的取向平行于x轴, 由方向向量(1,0,0)定义, 对应于第4, 5, 6个参数. 圆柱的取向在空间中是任意的.
10. Constrain rotations 约束旋转
对每种类型所有分子都可以约束其旋转, 这样它们在空间中具有一定的平均取向.
在structure...end structure节中, 约束旋转使用的关键词为
constrain_rotation x 180. 20.
constrain_rotation y 180. 20.
constrain_rotation z 180. 20.
每个关键词限制绕每个轴可能的旋转角度, 在180±20度(或其他值)范围内. 因技术原因, 绕z轴的单独旋转会与绕y轴的旋转完全相同(我们希望以后能修正这个问题). 约束三个旋转将会完全约束旋转. 注意, 要使你的分子取向平行于某个轴, 你需要约束分子相对于另外两个的旋转.
例如, 旋转约束分子使其沿z轴, 可使用下面的语句
constrain_rotation x 0. 20.
constrain_rotation y 0. 20.
注意, 这些旋转的定义是相对于用户提供的输入PDB文件中的取向, 因此, 为更易理解旋转, 将分子以合理方式进行取向是个好主意. 例如, 如果分子沿某一方向伸长, 在提供分子坐标时, 使其较大维度沿z轴取向更好.
周期性边界条件
用户经常要求程序支持立方和长方盒子的周期性边界条件(PBC). 我们计划将来实现这个功能. 同时, 我们建议一个简单的方法来解决这个问题: 如果你将要在一个例如100 Å的盒子中对体系进行模拟, 那么定义立方盒子约束时使用98 Å的盒子. 这样, 当使用PBC构建实际的模拟盒子时,不同映像彼此之间相隔2 Å, 如下图所示. 在边界处将会有空隙出现, 但进行能量最小化和预平衡时, 这些空隙会很快消失.

不同原子的不同半径
自15.133版本起, 在堆积过程中可以为不同原子定义不同的半径. 默认情况下, 在最终结构中, 所有原子彼此间的距离至少大于由容差参数tolerance定义的值. 在这种情况下, 可以认为每个原子的半径都是距离容差的一半. 使用全原子模型模拟分子体系时, 容差取2.0 Å通常是个不错的选择.
因此, 大多数时候, 你不需要这个选项. 那些需要堆积多尺度模型的用户才需要这个选项. 在多尺度模型中, 全原子和粗粒化的表示在同一个体系中被组合起来.
很容易为不同分子, 或同一分子内的不同原子定义不同的半径. 只要在分子类型的structure ... end structure节, 或原子选区的atoms ... end atoms节中添加radius关键词即可.
例如, 对下面的情况
tolerance 2.0
structure water.pdb
number 500
inside box 0. 0. 0. 30. 30. 30.
radius 1.5
end structure
水分子中的原子其半径为1.5. 注意, 这意味着水分子之间的距离容差为3.0.
另一方面,
structure water.pdb
number 500
inside box 0. 0. 0. 30. 30. 30.
atoms 1 2
radius 1.5
end atoms
end structure
水分子中只有原子1和2(按water.pdb文件中列出的顺序)的半径为1.5, 原子3的半径为1.0, 由容差2.0决定.
始终要记住, 距离容差是每对原子的半径之和, 半径越大, 堆积越难. 此外, 还要记住, 能量最小化和预平衡会处理实际的原子半径, Packmol仅仅是给出初始的坐标文件.
最后, 目前的约束由原子 中心 满足. 因此, 如果你使用大的原子半径, 你可能需要调整盒子, 球体等的大小, 这样, 所有原子才处于需要的区域内. 对标准的全原子模拟, 这通常不是问题, 因为其原子半径通常很小.
自动溶剂化大分子
Packmol发布中包含了一个solvate.tcl脚本, 可使用水和离子(Na+, Cl-)溶剂化大的分子, 通常是蛋白质. 给定生物分子的PDB文件, 使用下面的命令运行脚本
solvate.tcl PROTEIN.pdb
脚本会创建一个用于Packmol的输入文件packmol_input.inp, 使用这个文件运行Packmol
packmol < packmol_input.inp
大分子就会被15 Å的水壳层溶剂化, 同时会添加离子以保持体系的电中性, 并使体系处于生理NaCl浓度, 0.16M. 脚本自动选择的参数(水分子个数, 离子个数, 等等)通常很合理, 但这些参数也可以利用附加选项手动控制, 如下
solvate.tcl structure.pdb -shell 15. -charge +5 -density 1.0 -i pack.inp -o solvated.pdb
参数说明
structure.pdb: 要溶剂化的pdb文件(通常是蛋白质)15.: 溶剂化层的厚度. 可选参数, 默认值为15.+5: 体系需要中和的总电荷. 可选参数, 默认情况下程序会视His残基为中性, Arg和Lys所带电荷为+1, Glu和Asp所带电荷为-1. Na+和Cl-浓度会设定为最接近0.16M的值, 近似为生理溶度. 此外, 使用-noions选项不添加任何离子, 只添加水分子.1.0: 需要的密度. 可选, 默认为1 g/ml.solvated.pdb: 溶剂化后体系输出文件的名称, 可选, 默认使用solvated.pdb.pack.inp: 创建的packmol输入文件的名称, 可选, 默认为pack.inp.
当不使用任何参数运行solvate.tcl脚本时, 所有这些选项都会输出. 脚本同时还会输出盒子的大小以及建议使用的PBC尺寸.
控制PDB文件中的残基编号
由于Packmol会在一个新的PDB文件中创建一个或多个分子的副本, 有一些选项用来控制新分子的残基编号如何设定. 共有4个选项, 由resnumbers关键词指定, 此关键词可接受4个值, 0, 1, 2或3, 可插入每类分子的structure ... end structure节中. 每个选项说明如下:
resnumbers 0
这种情况下, 所有残基编号会对应于每类分子, 与原始pdb文件中的残基编号无关. 这意味着, 如果你堆积10个水分子和10个乙醇分子, 水分子的编号为1到10, 乙醇分子的编号也是1到10.
resnumbers 1
这种情况下, 会保持原始pdb文件中的残基编号不变. 这意味着如果你堆积10个含有5个残基的蛋白质, 残基编号会被保留, 因此, 在相同蛋白质的每个分子中, 等价残基的编号会重复.
resnumbers 2
在这种情况下, 结构中所有残基的残基编号会根据相同文件中前面列出残基编号按顺序编号. 这意味着, 这意味着如果你堆积10个含有5个残基的蛋白质, 残基编号会从1到50.
resnumbers 3
在这种情况下, 残基编号相应于文件中所有残基的序列号. 也就是, 如果你堆积一个含有150个残基的蛋白质, 外加10个水分子, 水分子的编号为从151到161.
例如, 此关键词也可以这样使用
structure peptide.pdb
number 10
resnumbers 1
inside box 0. 0. 0. 20. 20. 20.
end structure
_默认: 默认情况下, 对只有一个残基的结构使用0选项, 对多于一个残基的结构使用1选项.
链标识: 也可以修改PDB文件的链标识(chain). 默认情况下, 每类分子会被设定为chain. 此外, 可在每类分子的structure...end structure节中使用changechains, 这样链标识会交替取两个值(例如A和B). 如果分子是多肽, 这可能会有用, 因为拓扑构建程序有时会认为相同链的多肽必须有共价键连接. 通过交替改变链标识可避免这一点.
构建非常大的体系: 使用重启文件
自16.143版本起, 可以使用重启文件, 从多个互相独立的Packmol执行来构建体系. 要输出重启文件, 必须使用下面的关键词restart_to restart.pack, 其中restart.pack为要创建的重启文件的名称. 可以输出整个体系的重启文件, 如果关键词放于structure...end structure节外部的话, 也可以为体系的特定部分写出重启文件, 使用的语句类似下面这样:
structure water.pdb
number 1000
inside cube 0. 0. 0. 40.
restart_to water1.pack
end structure
这只会为水分子创建重启文件.
这些重启文件, 可以用于重新启动一个含更多分子的Packmol, 必须使用restart_fro关键词. 例如
structure water.pdb
number 1000
inside cube 0. 0. 0. 40.
restart_from water1.pack
end structure
新的输入文件可以包含其他分子, 像常规的Packmol输入文件一样, 这些水分子会和新的分子堆积在一起, 但从以前的运行开始. 这可用于, 例如一部分一部分地构建溶剂化的双层. 例如, 可以先构建双层, 然后添加不同的溶剂到双层, 而无须每次重新从头开始构建整个体系. 这也可用于添加一些分子到双层.
提示: 重启文件可用于重启更少相同类型分子的位置. 例如, 如果一个新分子被引入到以前的分子集合中(如双脂层), 你可以指示Packmol堆积更少的原始集合中的分子, 为新结构提供空间, 同时使用更多分子的原始重启文件. 也就是, 类似于上面例子中的restart_from water1.pack, 能够用于重启800个分子的位置.
收敛问题, 如何解决
有时Packmol无法找到一个适当的堆积结构. 这里我们给出一些提示, 以解决收敛问题:
- 查看已经得到的最佳解. 很多时候这些结构已经足够好了, 能够作为初始结构用于模拟
- 只使用每种类型的一些分子来优化. 例如, 不要使用20k个水分子, 而是使用300个, 看看它们是不是处于正确的区域中
- 如果你使用的分子非常大, 试着运行程序两次, 首先堆积这些分子, 然后使用得到的结构作为固定分子进行下次堆积, 也包括溶剂化. 当构建溶剂化的膜时, 这可能非常有帮助. 先构建膜然后使用它作为固定的分子来进行溶剂化.
-
你可以改变一些堆积过程的选项来加强优化过程
-
discale [实数]
此选项控制局部优化方法中实际使用的距离容差. 我们发现使用大的距离有时会有帮助. 例如, 试着将
discale设为1.5. -
maxit [整数]
这是每次循环中局部优化(GENCAN)的最大迭代次数. 目前的默认值是20, 改变此值收敛情况可能更好(或更差)
-
movebadrandom
Packmol的一种收敛策略是移动那些放置得不好的分子, 如果设定了这个选项, 分子将会被放置在盒子中新的随机位置上. 否则(默认)分子会被移动到堆积好的分子的附近位置. 当限制很复杂时, 使用此选项可能会有帮助, 但当结构很大时, 可能很差, 因为新的随机位置可能与那些重叠.
-
额外的输入选项和关键词
有一些输入选项, 一般的用户可能不敢兴趣, 但在特殊情况下, 这些选项可能会有用. 这些关键词可以添加到输入文件的任意位置.
- 在分子之间添加TER标识(AMBER使用此选项):
add_amber_ter -
增加盒子边长信息到输出的PDB文件中(GROMACS使用此选项):
add_box_sides 1.0.其中
1.0为可选的实数, 为每边增加的长度. 如果模拟盒子实际的边并不精确地等于体系中分子的最大最小坐标(通常, 如果你使用周期性边界条件, 你可能想要增加1.0来避免边界处的碎裂). -
增加最大体系的尺寸:
sidemax [实数] (如: sidemax 2000.d0)sidemax用于构建分子分布的初始近似. 如果你的体系非常大, 或者看起来被最大尺度切断了, 请增加sidemax. 体系的所有坐标必须处于-sidemax和+sidemax之间. 使用大致符合体系的sidemax, 可能会加速堆积计算. 默认值为1000.d0. -
改变随机数产生器的种子:
seed [整数] (如: seed 191917)使用-1会根据计算机时间自动生成种子
- 使用真正随机的初始点来最小化(默认选项是生成一个均匀密度的初始点, 固定分子之间没有重叠):
randominitialpoint - 改变每个循环中Gencan迭代的最大数目:
maxit [整数] - 改变循环的最大数目:
nloop [整数] - 改变输出文件写入的频率:
writeout [整数] - 将当前结构写入输出文件, 即便它比目前的最佳结构更差(仅用于检查目的):
writebad - 检查初始点, 仅用于测试目的, 仅构建初始近似, 写入输出文件, 然后退出:
check -
修改优化例程打印输出:
iprint1 [整数] 和/或 iprint2 [整数]整数必须是0 1 2 3
-
修改连接分胞方法的分格数目(技术):
fbins [实数]默认值为根号3
- 比较解析和有限差分的梯度. 仅仅用于测试的目的. 将比较结果写入
chkgrad.log文件:chkgrad
使用示例
尿素-水混合溶液
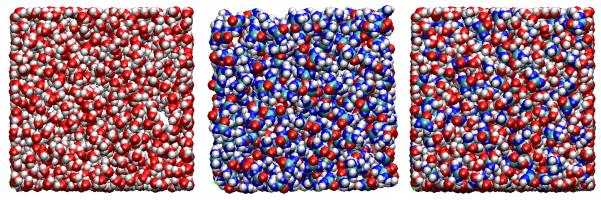
| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 | # 可以使用注释
# 尿素-水混合溶液
# 距离容差, 不同分子中原子间的最小距离2A
tolerance 2.0
# 输入输出文件的格式
filetype pdb
# 输出文件的名字
output mixture.pdb
# 1000个水分子, 400个尿素分子
# 盒子顶点坐标(x,y,z) 最小(0, 0, 0) 最大(40, 40, 40)
# 也即边长为40A的正方盒子
# 也可使用简单写法 inside cube 0. 0. 0. 40
structure water.pdb
number 1000
inside box 0. 0. 0. 40. 40. 40.
end structure
structure urea.pdb
number 400
inside box 0. 0. 0. 40. 40. 40.
end structure
|
水-四氯化碳界面上的甲状腺激素分子
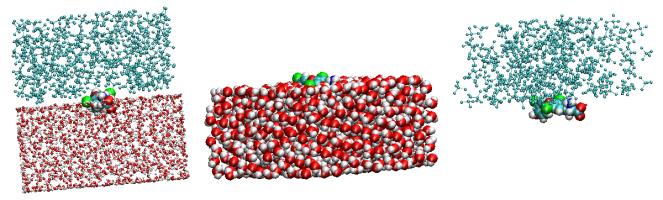
| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 | # 设定距离容差
tolerance 2.0
# 文件格式为简单的Molden-xyz格式
filetype xyz
output interface.xyz
# 1019个水分子, 处于20×39×39 的长方盒子中
# 盒子顶点坐标 最小(-20, 0, 0) 最大(0, 39, 39)
structure water.xyz
number 1019
inside box -20. 0. 0. 0. 39. 39.
end structure
# 199个四氯化碳分子, 处于与水相同大小的盒子内
# 但其盒子顶点x坐标从0开始, 最小(0, 0, 0) 最大(21, 39, 39)
# 这样在x=0处形成界面
structure chlor.xyz
number 199
inside box 0. 0. 0. 21. 39. 39.
end structure
# 甲状腺激素分子固定于界面处
# 其中心固定于盒子中心(0, 20, 20)
# 并旋转(pi/2, pi/2, pi/2) 使其看起来平行于界面
structure t3.xyz
centerofmass
fixed 0. 20. 20. 1.57 1.57 1.57
end structure
|
上下有水的双脂层

| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 | # 距离容差
tolerance 2.0
# 文件格式PDB(默认类型, 不写也可以)
filetype pdb
output bilayer.pdb
# 500个水分子, 处于双脂层下方
# 盒子 40×40×10, 顶点 (0, 0, -10)--(40, 40, 0)
structure water.pdb
number 500
inside box 0. 0. -10. 40. 40. 0.
end structure
# 500个水分子, 处于双脂层下方
# 盒子 40×40×10, 顶点 (0, 0, 28)--(40, 40, 38)
structure water.pdb
number 500
inside box 0. 0. 28. 40. 40. 38.
end structure
# 下方脂质层, 50个脂分子, 极性头部向下指向水盒子
# 盒子 40×40×14, 高度14比分子长度稍大
# 顶点 (0, 0, 0)--(40, 40, 14)
# 原子31和32属于极性头部, 被约束在z=2的平面以下
# 原子1和2为非极性端, 被约束在z=12的平面上
# 这样所有脂分子会沿z轴取向, 极性头部指向下方的水盒子
structure palmitoil.pdb
number 50
inside box 0. 0. 0. 40. 40. 14.
atoms 31 32
below plane 0. 0. 1. 2.
end atoms
atoms 1 2
over plane 0. 0. 1. 12.
end atoms
end structure
# 上方脂质层, 50个脂分子, 极性头部向上指向水盒子
# 与上面类似, 但取向相反
structure palmitoil.pdb
number 50
inside box 0. 0. 14. 40. 40. 28.
atoms 1 2
below plane 0. 0. 1. 16.
end atoms
atoms 31 32
over plane 0. 0. 1. 26
end atoms
end structure
|
内外有水的双层囊泡

| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 | tolerance 2.0
output spherical.pdb
filetype pdb
# 308个水分子处于半径为13的球中, 球心在原点
# 此水球处于囊泡内部
structure water.pdb
number 308
inside sphere 0. 0. 0. 13.
end structure
# 90个脂分子置于水球的周围
# 脂分子的极性头部要指向水, 因此原子37约束在半径为14的球内
# 非极性端要指向外部, 因此原子5约束在半径为26的球外
structure palmitoil.pdb
number 90
atoms 37
inside sphere 0. 0. 0. 14.
end atoms
atoms 5
outside sphere 0. 0. 0. 26.
end atoms
end structure
# 300个另外的脂分子形成新的层
# 非极性端指向球的内部, 这样它们可以与内层的非极性端相互作用
# 极性头部指向球的外部
structure palmitoil.pdb
number 300
atoms 5
inside sphere 0. 0. 0. 29.
end atoms
atoms 37
outside sphere 0. 0. 0. 41.
end atoms
end structure
# 最后, 17536个水分子形成溶剂化层
# 这些水分子处于前面定义的体系的外部(半径为43的球)
# 处于边长为95A的大盒子中
# 盒子顶点 (-47.5, -47.5, -47.5)--(47.5, 47.5, 47.5)
structure water.pdb
number 17536
inside box -47.5 -47.5 -47.5 47.5 47.5 47.5
outside sphere 0. 0. 0. 43.
end structure
|
水和离子溶剂化的蛋白质

| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 | tolerance 2.0
filetype pdb
output solvprotein.pdb
# 蛋白质的中心固定于盒子中心, 不旋转
structure protein.pdb
number 1
fixed 0. 0. 0. 0. 0. 0.
centerofmass
end structure
# 16500个水分子置于包含蛋白质的球内部
# 球心位于原点, 半径为50A
structure water.pdb
number 16500
inside sphere 0. 0. 0. 50.
end structure
# 离子位置与水分子相同
# 这样才可以处于水球内并溶剂化蛋白质
structure CLA.pdb
number 20
inside sphere 0. 0. 0. 50.
end structure
structure SOD.pdb
number 30
inside sphere 0. 0. 0. 50.
end structure
|
网络资料
评论
- 2016-09-23 01:24:38
逗比大懒猪这东东我以前也玩过 做混合溶剂, 是对付amber的. 做成一个混合溶剂盒子用于日后溶剂化, 但盒子的边界总感觉不均匀 - 2016-09-23 14:59:17
Jerkwin是这样的, 因为构造时没有考虑PBC的原因, 但作为初始构型足够了.
