- 发布: 2016-10-11 15:53:25; 翻译: 曾宪阳(吉林大学, 建议或问题可联系1543976771@qq.com); 校对: 李继存
如果你是第一次练习此教程, 我们建议你先阅读一下Martini的脂质双分子层教程.
溶液中的蛋白
遵循Martini粗粒化粒子的定义规范, 在粗粒化的蛋白模型中, 1个粗粒珠子由4个非氢重原子组成. 根据残基类型的不同, 每个氨基酸残基有1个骨架珠子和0-4个侧链珠子组成. 蛋白质的二级结构影响每个残基的珠子类型以及键长, 键角, 二面角参数[1]. 值得注意的是, 尽管蛋白的三级结构可以任意改变, 局部的二级结构是预先定义好的, 从而在整个模拟过程中保持不变. 因此涉及二级结构改变的构象变化是无法使用Martini粗粒化蛋白进行研究的.
建立粗粒化蛋白模拟的三个基本步骤:
- 将全原子蛋白模型转换为粗粒化蛋白模型
- 生成与粗粒化模型匹配的拓扑文件
- 在所需要的环境中对蛋白进行溶剂化
前两步使用martinize.py脚本完成, 其最新版本可以在这里下载. 最后一步使用GROMACS中的工具和/或特定脚本完成. 在这部分教程中, 我们默认大家熟悉GROMCS的一些基本知识, 所以不会明确地列出所有的命令. 请参考之前的Martini脂质基础教程和/或GROMACS手册.
本教程第一个模块的目标是说明常规的工作流程和策略, 运用标准的Martini粗粒化方法建立泛素在水中的粗粒化模拟. 第二个模块则展示二级结构的变化, 在一些特定的蛋白中会出现这种状况, 我们使用的例子是HIV-1蛋白酶, 当然我们也提供了稍有不同的方法来避免这种情况发生. 最后一个模块是进行膜环境中的KALP多肽的粗粒化模拟.
可溶性蛋白: Martini说明
下载的文件为ubiquitin.tgz, 包含整理过的该模块所需的命令集. 你可以使用它们去对照检查自己的操作. 这个文件解压后会释放到soluble-protein目录, 本模块的文件在ubiquitin/martini目录下. 创建你自己的工作目录亲自尝试一下教程的内容. 对你需要下载文件会给出一些说明, 等等. 你不需要任何文件就可以开始. 现在进入到你自己的工作目录.
得到泛素(1UBQ)的原子结构文件后, 你需要将它转换为粗粒化结构, 然后准备好相对应的Martini拓扑文件. 当这一步完成后, 可以对粗粒化结构进行能量最小化, 溶剂化和模拟. 需要的步骤大致如下:
-
下载从PDB蛋白库中下载1UBQ.pdb
wget http://www.rcsb.org/pdb/files/1UBQ.pdb.gz gunzip 1UBQ.pdb.gz -
下载好的pdb结构可以直接使用
martinize.py脚本(可以在这里下载)进行处理, 从而生成粗粒化的结构和拓扑文件. 具体可以参考帮助(即运行martinize.py -h)来查看可用选项. 提示, 对使用Martini进行模拟的任何体系, 如下操作都是合理的: 在平衡阶段对(骨架)使用位置限制以弛豫侧链及其与溶剂的相互作用, 可能会有帮助; 我们可通过martinize.py的选项来生成一个涉及位置限制原子的列表. 最终的命令大致如下:martinize.py -f 1UBQ.pdb -o system-vaccum.top -x 1UBQ-CG.pdb -dssp /路径/dssp -p backbone -ff martini22下载文件: martinize.py, system-vaccum.top, 1UBQ-CG.pdb
我们使用了2.2版本的力场. 当使用
-dssp选项时, 你需要先安装dssp程序, 用于根据蛋白质结构确定其骨架的二级结构. 它可以从CMBI网站下载dssp程序. 作为另一种选择, 你可以自己准备一个包含所需二级结构的文件并提交给脚本:martinize.py -f 1UBQ.pdb -o system-vaccum.top -x 1UBQ-CG.pdb -ss <你的文件> -p backbone -ff martini22注意
martinize.py脚本可能与老版本的python不兼容! 并且与新近的python 3.x版本也不兼容. 我们确知它与2.6.x版本, 2.7.x版本兼容, 不与2.4.x版本兼容. 如果你测试了处于兼容和不兼容版本之间的任何版本, 我们很愿意知道你的测试结果. -
如果以上步骤进行得很好, 脚本会生成3个文件: 一个粗粒化结构(
.gro/.pdb; 图1), 一个主拓扑文件(.top)和一个蛋白拓扑文件(.itp). 为进行模拟你还需要其他两个文件: Martini拓扑文件(martini_v2.x.itp, 如果你指定了2.2版本, 你需要相应的文件), 还需要一个运行参数文件(.mdp). 你可以从Martini网站的例子或蛋白教程中获得它们. 需要时, 别忘了调整设置!`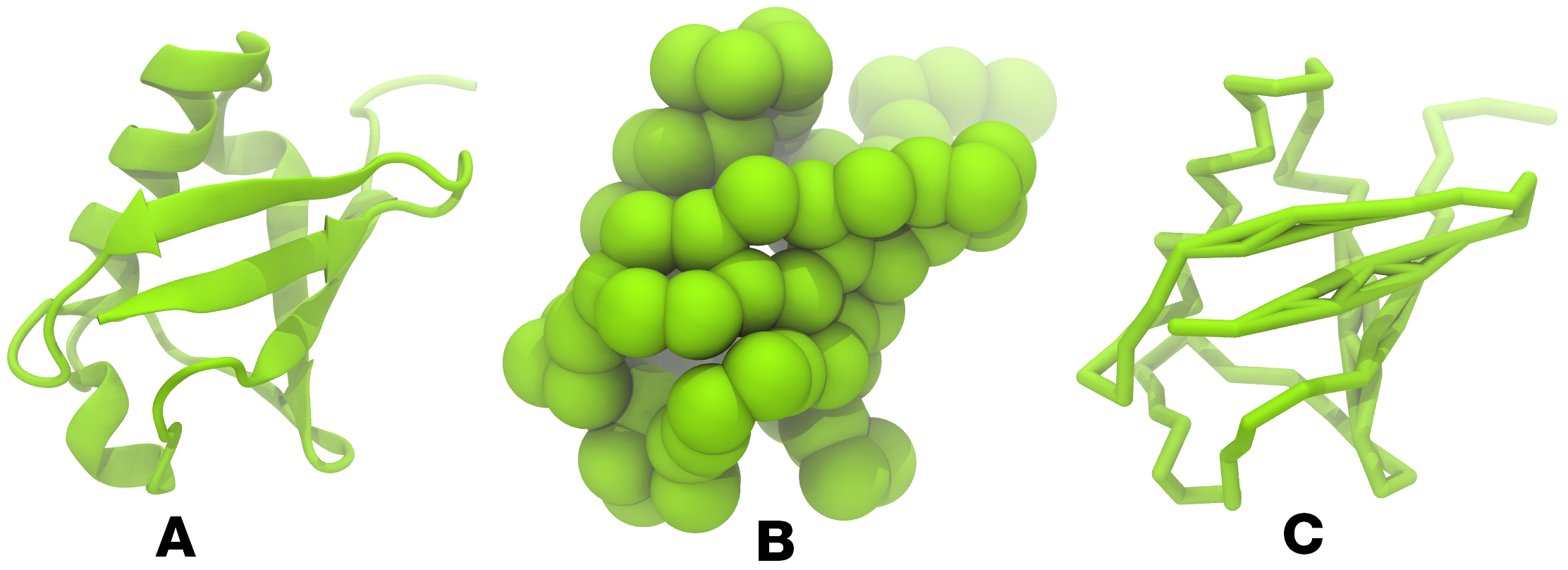
图1 泛素的不同表示形式. A) cartoon形式的原子结构. B) 粗粒化珠子形式的骨架 C) licorice形式下的粗粒化蛋白骨架
-
做一个简短的真空能量最小化(约10步就足够了). 在你用
grompp生成输入文件之前, 你需要检查拓扑文件(.top)是否引用了正确的Martini参数文件(.itp). 如果引用的.itp文件不正确, 你需要修改其中的引用语句. 此外, 你还必须生成一个盒子, 并指定其尺寸大小, 例如可使用editconf来完成. 你需要保证盒子足够大以避免蛋白的周期映象彼此靠得太近, 而且边长要大于模拟所用截断半径的两倍. 试着使蛋白边缘到任意盒子边缘的距离至少是1 nm. 然后, 复制例子中的参数文件, 修改相关设置做一次能量最小化. 现在你已经可以做预处理和能量最小化了:editconf -f 1UBQ-CG.pdb -d 1.0 -o 1UBQ-CG.gro grompp -f minimization-vaccum.mdp -p system-vaccum.top -c 1UBQ-CG.gro -o minimization-vaccum.tpr mdrun -deffnm minimization-vaccum -v下载文件: minimization-vaccum.mdp, system-vaccum.top, 1UBQ-CG.gro, minimization-vaccum.tpr
-
使用
genbox填充溶剂(可以在这里下载一个平衡好的水盒子, 文件名为water.gro. 在下面命令中它被保存为water-box-CG_303K-1bar.gro). 确保盒子的尺寸足够大(即保证蛋白周围有足够的水, 以避免因周期性边界条件产生假象), 与此同时还要记得在溶剂化过程中使用较大的范德华距离以避免崩溃, 命令范例:genbox-cp minimization-vaccum.gro -cs water-box-CG_303K-1bar.gro -vdwd 0.21 -o system-solvated.gro下载文件: minimization-vaccum.gro, water-box-CG_303K-1bar.gro, system-solvated.gro
-
对填充溶剂后的体系做一个短的能量最小化和位置限制模拟. 既然
martinize.py脚本已经生成了一个位置限制文件(-p选项的作用), 你唯一要做的就是在参数文件(.mdp)中标明define = -DPOSRES, 然后在体系的拓扑文件(.top)中加入适当数量的水珠子(分子名称为W); 这个数目可以在genbox命令的输出中看到.grompp -f minimization.mdp -c system-solvated.gro -p system.top -o minimization.tpr mdrun -deffnm minimization -v grompp -f equilibration.mdp -c minimization.gro -p system.top -o equilibration.tpr mdrun -deffnm equilibration -v下载文件: minimization.mdp, system-solvated.gro, system.top, minimization.tpr, equilibration.mdp, minimization.gro, system.top, equilibration.tpr
-
开始成品模拟(不含位置限制!). 如果模拟体系崩溃了, 可能你需要增加预平衡的步数. 图2展示了一条示例轨迹.
grompp -f dynamic.mdp -c equilibration.gro -p system.top -o dynamic.tpr mdrun -deffnm dynamic -v下载文件: dynamic.mdp, equilibration.gro, system.top, dynamic.tpr

图2 粗粒化泛素(绿色珠子)和溶剂(粗粒化的水, 蓝色珠子)的20 ns模拟.
-
重要提示! 这个分子可以进行哪些分析? 先在
vmd中打开看一下.
可溶性蛋白: 弹性网络方法
本教程第二个模块的目标是展示, 弹性网络方法如何与Martini模型联合使用, 以更忠实地保持蛋白质的二级, 三级和四级结构, 而同时不牺牲蛋白的真实动力学性质. 我们提供两套可选方案. 但请诸位明白, 目前这是一个活跃的研究领域, 还没有固定不可替代的黄金法则. 第一套方案是基于标准Martini拓扑生成一个简单的弹性网络, 第二套方案是使用EINeDyn网络. 第二套方案对Martini力场的修改很多, 所以其自身是一个不同的力场. 它的优点是已有研究很好地描述了方法的行为[2]. 这两种方案都可以使用matinize.py脚本来进行设置, 我们将在下面对它们做简短的介绍.
我们建议, 第一步先做一个不含弹性网络的纯粗粒化蛋白, 然后对比一下应用弹性网络后相同的蛋白会发生哪些结构变化. 但需要注意, 你大概需要几十或几百纳秒的模拟时长才能观察到结构的主要变化. 在我们提供的文件中有模拟好的范例.
Martini
- 使用HIV-1蛋白酶(
1A8G.pdb)重复前一练习的1-7步. - 观察模拟结果, 尤其是蛋白的结合口袋部分: 它是保持关闭的还是开放的? 整个蛋白的结构是否变化?
Martini+弹性网
为维持蛋白的高级结构, 第一套方案的作法是, 在标准的Martini拓扑中增加额外的简谐键, 这些键位于未成键的珠子之间, 基于截断距离来设定. 注意在标准的Martini中, 长的简谐键已被用于蛋白拓展片段的二级结构上了. 如果你使用了-elastic选项, martinize.py脚本将会在骨架珠子之间生成简谐键. 可以调整弹性键(例如: 使力常数与距离有关, 上调或下调截断半径, 等等)使得蛋白行为更加合理. 为了找到合适的参数, 唯一的方法就是尝试不同的选项, 并将蛋白的行为与全原子模拟结果或实验数据(如核磁共振, 等等)做比较. 这里我们只使用基本的参数来对其原理进行展示.
-
如前所述, 使用
martinize.py脚本生成粗粒化结构和拓扑. 对于弹性网, 我们加入额外的选项:martinize.py [...] -elastic -ef 500 -el 0.5 -eu 0.9 -ea 0 -ep 0-elastic选项将弹性网络应用到粗粒结构上,-ef 500将弹性键力常数设置为500 kJ mol-1 nm-2,-el 0.5和-eu 0.9设置弹性键截断距离的下限和上限分别为0.5和0.9 nm, 并设置键强度不受键长的影响(弹性键衰减因子和衰减阶数的默认值为-ea 0和-ep 0). 在.itp文件中, 弹性网络通过一个#ifdef语句定义, 而且默认是开启的(在.top文件或.itp文件中加上#define NO_RUBBER_BANDS可以关闭弹性网络). 注意martinize.py脚本并不会在第i→i+1, i→i+2个骨架键之间生成弹性键, 因为它们之间已经被键和键角连在一起了(请参看图3).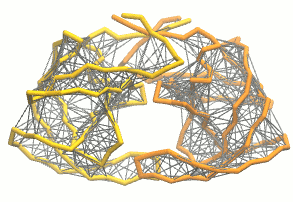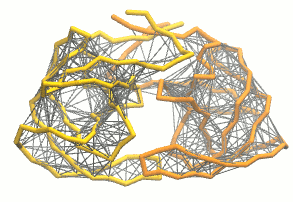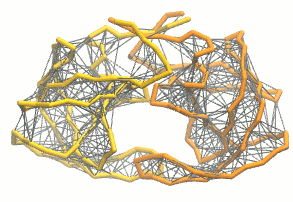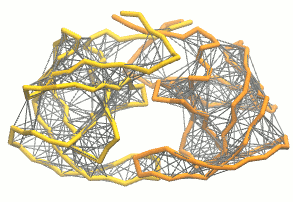
图3 弹性网络(灰色)应用在HIV-1蛋白酶的两个单体上(分别为黄色和橘色)
-
像之前的4-7步那样, 进行一个成品模拟. 注意我们对蛋白增加了一个附加级别的约束; 因此可能需要一些附加的弛豫步骤(例如, 使用位置限制和更小的时间步长进行平衡).
EINeDyn
弹性网络与Martini联合使用的第二套方案更强调接近PDB文件中原有的蛋白整体结构而不是标准Martini模型的结构. 与标准方法(即我们之前所做的练习)的主要不同是, 在骨架珠子之间使用整体的弹性网络来保持蛋白的整体构象, 而不是依赖键角/二面角势能以及局部弹性键. 骨架珠子的位置也与之前的稍有不同: 在标准Martini模型中, 骨架珠子位于多肽平面的质心, 但在ElNeDyn中位于Cα原子. martinize.py脚本会自动设定这些选项, 并为弹性网络设定正确的参数. 由于在ElNeDyn弹性网络中必须调整弹性键的强度和截断的上限, 因此这些选项可以手动设定(-ef和-eu). 注意这些参数都已经被广泛地研究过了, 最优设定是500 kJ mol-1 nm-2和0.9 nm[2].
-
和先前一样, 使用
martinize.py脚本生成粗粒化结构和拓扑.martinize.py [...] -ff elnedyn22 -
如前步骤, 运行成品模拟.
现在对于同一个蛋白, 你已经进行三个模拟, 各自使用了不同类型的弹性网络. 如果你不想等待模拟完成, 压缩包提供了一些我们运行过的轨迹. 它们中有一个或许能满足您对于结构和动力学的需要. 否则的话, 还有无穷多的方法来调整弹性网络!
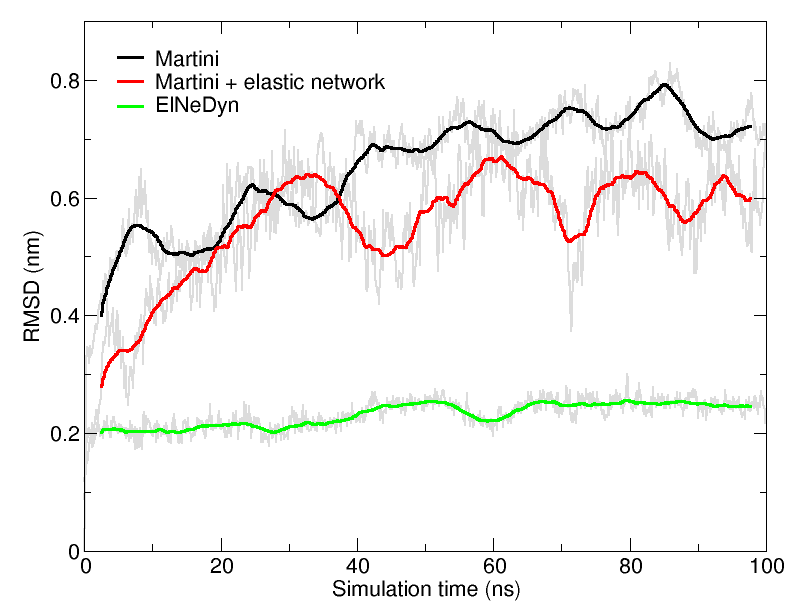
图4 三种不同方法下HIV-1蛋白酶骨架的RMSD(低于0.3 nm的RMSD一般认定为是合理的)
比较前面三种不同方法下蛋白行为微小差异的一个简单方法就是观察骨架在模拟中的偏差/波动情况(如果可能, 与全原子模拟的结果相比较). RMSD(图4)和RMSF可以通过GROMACS的工具进行计算. vmd也提供的一些好用的工具来计算这些性质, 但需要一些技巧才能适用于粗粒化体系(对粗粒化结构, vmd不能识别标准关键字).
进阶: 膜蛋白
下载该模块所需的所有文件(适用于旧式的脂质拓扑文件)
下载该模块所需的最少文件并自己使用脂质拓扑来生成需要的全部文件
在本蛋白教程的最后一个模块中, 我们加大体系的复杂度, 将蛋白嵌入脂质双分子层中(使用旧式脂质双分子层的教程见这里, 使用脂质组的教程见这里). 我们将观察一下多肽KALP的二聚体在DPPC膜中的倾斜程度. 粗粒化结构(kalp.gro)和拓扑(kalp.itp)在压缩包中, 你不需要再重复上面的1-4步粗粒化步骤. 我们从第5步开始, 因为我们会用不同的方式来溶剂化蛋白质. 具体如何执行这一步有很多不同的方法. 下面是一个不完全的列表, 方法的复杂程度依次递减:
- 双分子层自组装动力学, 像脂质教程第一个模块中的一样, 会使得KALP自身嵌入双分子层(图5).
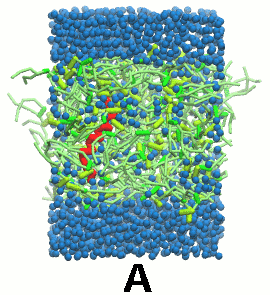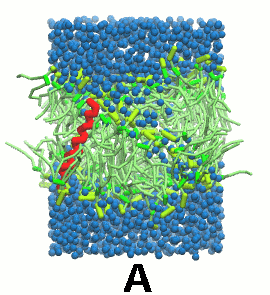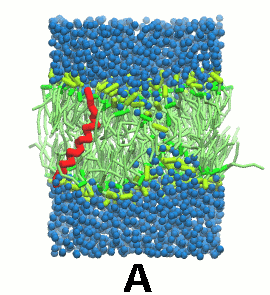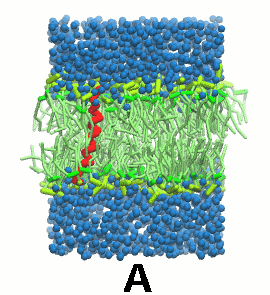
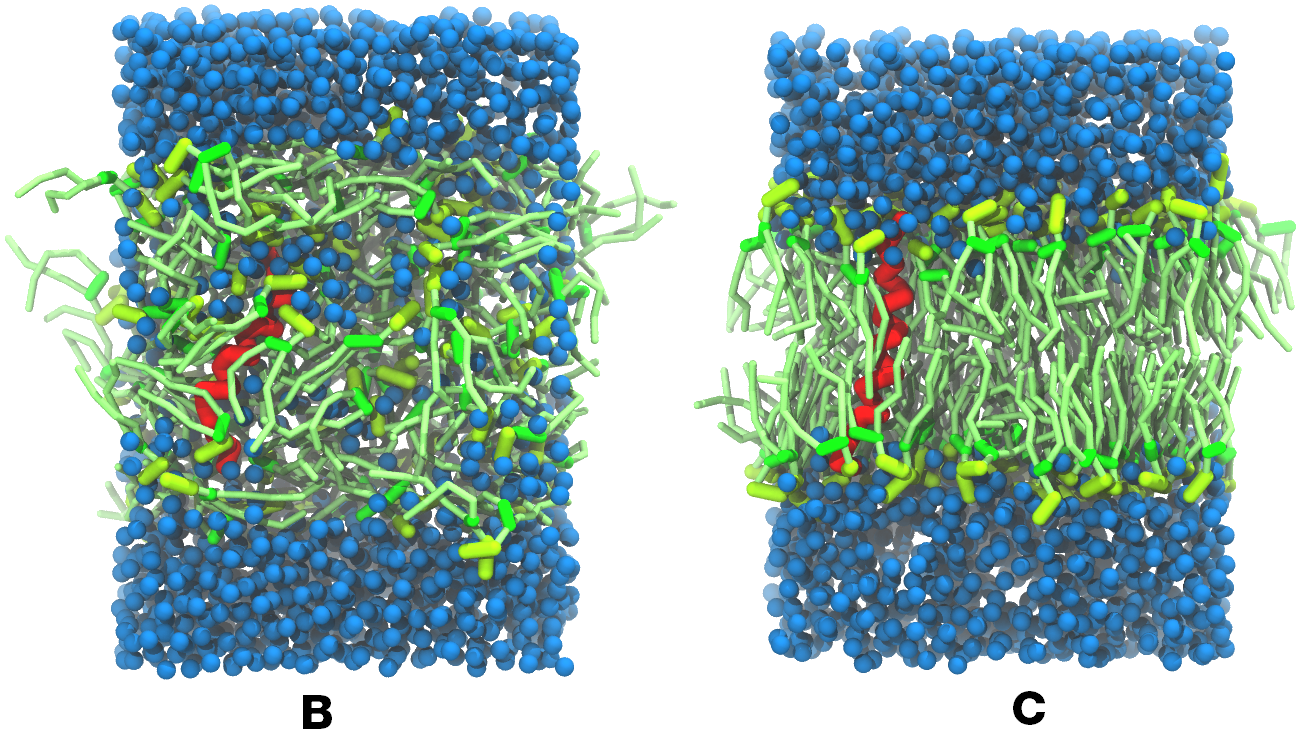
图5 A) KALP蛋白嵌入自组装(4 ns)的DPPC双分子层(绿色). B) 混合着脂质, 溶剂(水, 蓝色)和KALP(红色)的初始的混乱态. C) 模拟的最终构象.
- 如果已经有了一个与所需盒子大小匹配的稳定的DPPC膜,
genbox可用于将蛋白嵌入双分子层中(需要预先将蛋白居中以保证能嵌入膜中). - 使用
insane.py脚本, 可以在这里下载.
最后一种方法可以将蛋白嵌入任意成分的双分子层中. 既简单又直接, 我们推荐也在这里运用这种方式.
-
insane.py脚本的语法和之前的脚本类似, 可以通过insane.py -h来查看可用选项. 让我们来看一个实际的例子:insane.py -f kalp.gro -o system.gro -p system.top -pbc square -box 10,10,10 -l DPPC -center -sol W下载文件: insane.py, system.gro
上面的命令将会建立一个完整的体系, 包括一个边长为10 nm的DPPC正方双分子层, 中央嵌入KALP蛋白, 并使用标准粗粒化的水分子对它们进行溶剂化. 关于
insane.py的更多功能可在其他几个教程中找到, 尤其是建立复杂双分子层的教程. (使用旧式脂质的见这里). -
我们继续并开始运行成品模拟. 提醒大家, 操作包括: (1) 编辑
system.top文件指定你想使用的Martini版本(KALP拓扑在2.1版本中提供), 并注明需要引用的拓扑; (2) 下载或复制你需要的Martini版本, 脂质库中DPPC脂质的拓扑; (3) 所有涉及的分子都要使用正确的命名; (4) 下载或复制用于能量最小化, 预平衡和成品模拟所需要的.mdp文件, 如果需要, 编辑它们(模拟双分子层时最好使用半各项异性压力耦合, 你也许需要将不同的组分开耦合到热浴); (5) 运行最小化和预平衡. -
生成一个新的体系, 其中膜的厚度小一些(使用不同类型的膜, 比如DLPC). 观察膜的厚度对蛋白跨膜螺旋的倾斜程度的影响; 与之前的模拟结果进行比较.
-
将之前的盒子在某一方向上加倍(使用
genconf工具), 然后再次运行模拟. 观察蛋白二聚体的构象(平行和反平行的倾斜). 注意可能需要多次模拟才能观察到这两种情况! -
选择你最喜欢的方向, 将最后的构象进行逆映射(逆映射教程见这里). 然后使用全原子模型模拟这个体系, 仔细观察KALP之间相互作用的细节.
教程中使用到的脚本和工具
- GROMACS: http://www.gromacs.org/
martinize.py: 点击这里下载insane.py: 点击这里下载
参考文献
- L. Monticelli, S.K. Kandasamy, X. Periole, R.G. Larson, D.P. Tieleman, S.J. Marrink. The MARTINI coarse grained forcefield: extension to proteins. J. Chem. Theory Comput., 4:819-834, 2008.
- X. Periole, M. Cavalli, S.J. Marrink, M.A. Ceruso. Combining an elastic network with a coarse-grained molecular force field: structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput., 5:2531-2543, 2009.
