- 2017-08-20 14:13:12 初稿
- 2018-05-03 19:04:27 修订
- 2020-11-11 11:57:52 分组, 高精度, GMX2019.6测试通过
上一篇博文 【整理】使用分子动力学模拟红外光谱中整理了根据分子动力学模拟计算红外光谱的理论基础知识, 既然已经知道了计算方法, 剩下的就是如何实现了.
实际上只要有了体系偶极矩的演化数据, 计算其自相关函数, 再计算自相关函数的傅立叶变换是个很常见的需求, 很多程序和软件都可以实现. 如果你编程娴熟, 完全可以自己写代码进行分析. 但大多数人不具备这个能力, 所以我还是建议尽量利用GROMACS自带的分析工具达到目的. 这符合我一贯的主张: 尽量组合已有的工具实现需求, 同时注意减少工具链的长度.
查阅GROMACS的程序文档, 我们可以找到几个能进行自相关函数或光谱/功率谱计算的工具, gmx analyze, gmx dipoles, gmx dos, gmx velacc. 这些工具大都可以用来做自相关分析, 但关注的重点不同. 其中最通用的是gmx analyze程序, 最接近我们需求的是gmx velacc, 它可以做速度自相关函数, 并且有一个-os选项可以直接给出光谱, 而且这个工具可以通过使用组索引文件分析任意组的光谱, 便于指认光谱. 但这个工具只支持二进制的trr轨迹文件, 而不支持gro格式的轨迹, 而且计算结果是速度v自相关函数的光谱. 根据文献 Vishal Agarwal, George W. Huber, W. Curtis Conner, Scott M. Auerbach; J. Chem. Phys. 135(13):134506, 2011; 10.1063/1.3646306 的说法, 我们需要计算的是电流矢量vq自相关函数的光谱. 所以, 我们首先需要将trr中每个粒子的速度乘上其电荷, 以vq代替trr中的v. 要达到这一目的, 我们首先需要获知体系中每个粒子的电荷. 如果对电荷的精度要求不高, 可以使用gmx editconf -f *.tpr -mead *.pqr输出体系tpr对应的pqr文件, 其中包含了每个粒子的电荷值. 如果需要每个粒子电荷值的精确值, 需要自己对tpr文件中的电荷信息进行分析. 但由于tpr是二进制格式, 不易直接处理, 所以简单点的方法是先使用gmx dump -s *.tpr将其转换为普通的文本格式, 再分析其中的电荷信息. Sobereva在文章将GROMACS的原子电荷信息读入VMD的方法中对这个问题进行了说明, 并给出了一段相应代码, 但那段代码是Fortran写的, 用起来不大方便, 所以我使用一段bash代码实现这个功能.
知道了每个粒子的电荷值之后, 我们需要将每个粒子的速度v乘上其电荷值q, 得到每个粒子的vq值, 并以vq值替代trr中的v值. 理想的做法是直接读入trr, 然后输出trr. 如果你有能力使用编译语言直接读取和输出trr文件, 效率当然很好, 但这种方法对编程水平要求比较高, 实现代码也没那么容易写, 牵涉到很多麻烦的事情. 因为trr是二进制格式, 要读写的话, 必须借助GROMACS的库才可以完成. 只要轨迹没有达到上百G, 使用脚本处理起来问题不大, 而且更灵活. 所以, 在这里我们采用间接办法, 先将trr转成gro文件, 在gro文件中完成速度值的替换, 然后再将gro文件转换成trr文件, 这就避免了直接读写trr二进制文件的麻烦事.
首先将不易读写的trr文件转换为容易读写的gro文件, 然后将其中的速度乘上前面得到的电荷值, 得到新的gro文件, 再将替换后的gro文件转换为trr文件.
最后我们就可以使用替换速度值后的trr文件计算光谱了.
代码
见gmxtool.
说明
gmx velacc在计算光谱时并没有对自相关函数进行任何前处理, 如均值零化, 加窗, 滤波等, 也没有对得到的光谱进行任何后处理, 如平滑, 插值, 滤波, 量子校正等. 所以其数据可能与其他程序或软件给出的数据不同.- 所得光谱的分辨率dF是由时间长度决定的, 根据采样定理, dF>2/T, T是总时间. 由此我们知道, 如果要获得更高分辨率的光谱, 需要增长模拟时间. 若需要的光谱分辨率为1 cm^-1, 那么总时间长度必须大于66 ps, 再考虑到相关函数计算时大多只能得到总长度一半的有效数据, 那么总的时间长度还要加倍, 需要140 ps左右. 这样的模拟时间对经典分子动力学来说不算长, 但对从头算或第一原理动力学来说不算短.
- 所得光谱的最高频率是由时间间隔决定的, maxF<1/(2dt), dt为时间间隔. 一般认为红外光谱的范围为400-4000 cm^-1, 因此只要时间间隔小于4 fs即可满足要求.
- 根据相关函数计算光谱并非只有傅立叶变换一种方法, 另一种常用的方法是最大熵方法. 这种方法对数据的要求低于傅立叶变换, 但仍满足采样定理.
- 要计算光谱, 模拟时最好不要使用刚性模拟或者约束, 否则的话, 所得光谱只包含低频部分, 而缺失了高频部分的细节.
- 仔细查看
velacc计算相关函数的方法, 发现并不是先计算体系整体的电流矢量, 然后计算其相关, 而是对每个粒子的电流矢量计算相关, 然后进行平均, 因此, 如果只是简单地将原子速度乘上电荷再写回trr, 所得的光谱严格来说是不对的. 简单的解决方法是将整体的电流矢量写入单个原子的速度中. 上面的代码使用了这种方法. - GROMACS自2016版本起
trjconv输出gro时不再支持-ndec选项, 说明见Removed variable-precision .gro writing及其讨论. 但读入gro时仍支持变精度格式, 所以上面的代码中使用这种方法保持gro精度. 更好的方法是换用g96格式, 或者自己编写程序输出trr轨迹.
辞不尽言, 言不尽意, 惟意惟一, 愤启悱发. 如此吧.
示例
以SPC柔性水模型为例. 初始构型采用GROMACS自带spc216.gro, 进行NPT模拟, 时间步长2 fs, 模拟时间100 ps. 前50 ps预平衡, 后50 ps分析. 由于总时间有限, 所以所得光谱分辨率不是很高, 但作为示例足够了.
- vq的自相关函数
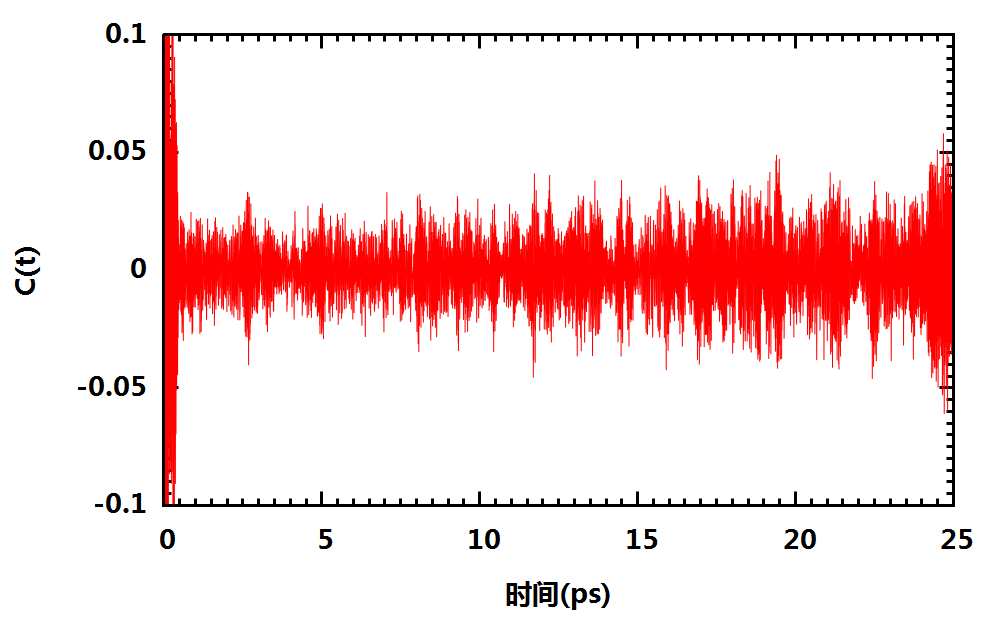
- 体系的红外光谱
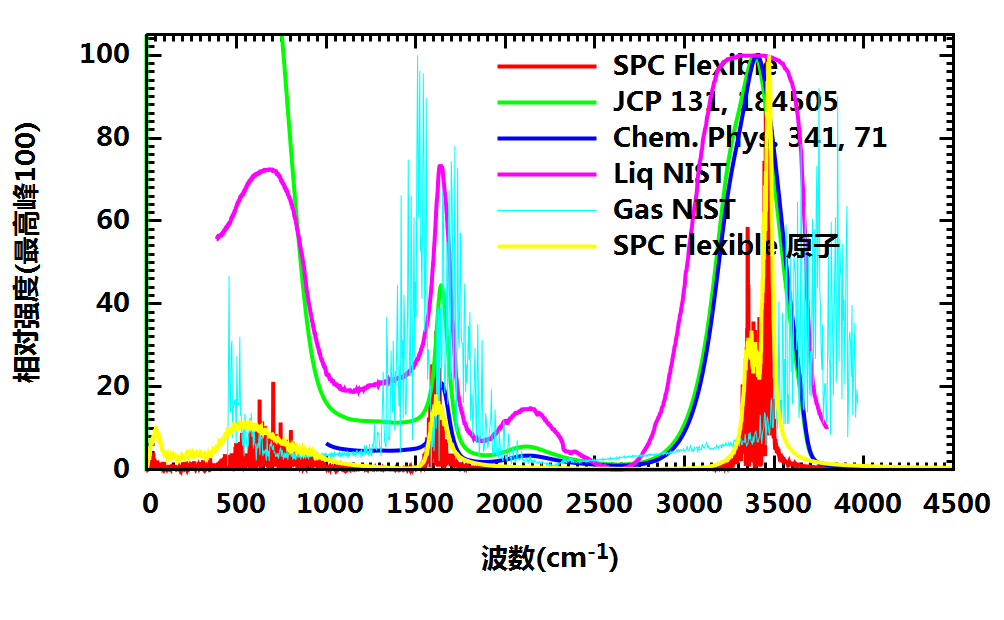
对比的实验光谱
- JCP 131, 184505: Jean-joseph Max, Camille Chapados; J. Chem. Phys. 131(18):184505, 2009; 10.1063/1.3258646
- Chem. Phys. 341, 71
- Liq NIST: NIST 液态水红外光谱
- Gas NIST: NIST 气态水红外光谱
参考
- Vishal Agarwal, George W. Huber, W. Curtis Conner, Scott M. Auerbach; J. Chem. Phys. 135(13):134506, 2011; 10.1063/1.3646306
- Jean-joseph Max, Camille Chapados; J. Chem. Phys. 131(18):184505, 2009; 10.1063/1.3258646
- Matej Praprotnik, Dušanka Janežič, Janez Mavri; J. Phys. Chem. A 108(50):11056-11062, 2004; 10.1021/jp046158d
- Timo Marcel Daniel Graen; Anharmonic Infrared Spectra From Short Qm/mm Simulations
- 王程超, 谭建宇, 杨家跃, 刘林华; 水和重水红外吸收光谱的 Car-Parrinello 分子动力学模拟; 科学通报, 60(31) 3014-3020, 2015
