- 原始文档: AMBER ADVANCED TUTORIAL 3: MM-PBSA
- Perl Version By Ross Walker & Thomas Steinbrecher
- Python Version By Dwight McGee, Bill Miller III, & Jason Swails
- 2018-01-28 06:39:37 翻译: 袁媛; 修订: 李继存

本教程使用mm_pbsa脚本计算RAS-RAF蛋白复合物的结合能, 并对计算中的每一步进行解释说明. 同时教程还包括了对如何使用mmpbsa_py脚本进行这种计算的说明.
AMBER高级教程A3: MM-PBSA
- AMBER高级教程A3: MM-PBSA

MM-PBSA方法可用于计算蛋白-配体复合物间的结合自由能, 其中配体可以是蛋白, 也可以是小分子, 多肽等等. 在本教程中, 我们将使用MM-PBSA方法计算两个蛋白结合的结合能.
MM-PBSA方法及其互补的MM-GBSA方法的总体目标是比较同一分子两种不同的溶剂化构象的自由能, 或者计算两个状态之间的自由能差, 这两个状态最通常的情况是代表两个溶剂化分子的结合和未结合状态.
\[[A]_{aq} +[B]_{aq} \iff [A^* B^*]_{aq^*}\]理想情况下, 我们想直接计算如下图所示的结合自由能:

然而, 在这些溶剂化状态的模拟中, 大部分能量贡献将来自溶剂-溶剂相互作用, 并且总能量的波动将比结合能大一个数量级, 因此结合能的计算需要非常长的时间才能收敛. 因而更有效的方法是按照下面的热力学循环将计算划分开来:

显然, 从上图可以看出结合自由能 $\D G_\text{bind,solv}$ 可以通过下式计算:
\[\D G_\text{bind,solv}^0=\D G_\text{bind,vacuum}^0+\D G_\text{solv,complex}^0-(\D G_\text{solv,ligand}^0+\D G_\text{solv,receptor}^0)\]在MM-PBSA方法中, 对上述结合自由能的不同贡献以不同方式进行计算:
- 对三种状态的溶剂化自由能是通过求解线性化的泊松-波尔兹曼(Poisson-Boltzman)方程或广义波恩(Generalized Born)方程(给出了静电相互作用对溶剂化自由能的贡献), 并添加表示疏水贡献的经验项来计算的.
- $\D G_\text{vacuum}$ 是通过计算受体和配体之间的平均相互作用能, 并且在必要或需要时考虑受体与配体结合时熵的变化来得到的:
-
熵变对于 $\D G_\text{vacuum}$ 的贡献可以通过对三个物种(即两个蛋白和蛋白-蛋白复合物)进行简正模式分析获得. 但实际上如果只是需要比较具有相似熵的状态, 比如两个配体结合到同一个蛋白, 那么熵对于 $\D G_\text{vacuum}$ 的贡献可以忽略. 这样做的原因在于简正模式分析计算非常费时, 而且一般存在较大的误差, 会在结果中引入显著的不确定性.
-
受体与配体间的平均相互作用能通常是通过对不相关的快照(snapshots)系综进行计算得到的, 这些快照来自一段平衡的分子动力学模拟.
在本教程中, 我们将演示使用Amber和AmberTools自带的MM/PB(GB)SA脚本自动执行所有必要步骤, 对蛋白-蛋白复合物(RAS和RAF)和蛋白-配体复合物(雌激素受体和雷洛昔芬)的结合自由能进行计算, 计算时MM-GBSA和MM-PBSA采用串行和并行两种模式. 此外, 我们还将演示使用脚本进行丙氨酸扫描和简正模式熵计算. 原则上, 上述结合自由能的计算需要对复合物以及两种单独蛋白质进行三次独立的MD模拟. 不过, 通常我们近似地认为, 在结合时不会发生显著的构象变化, 从而可以从单一轨迹获得所有三个物种的快照, 这称为”单轨迹方法”. 本教程就采用这种方法.
1. 构建初始结构, 运行模拟获得平衡体系
在模拟中我们将要建模的体系是人类H-Ras蛋白与C-Raf1的Ras结合结构域之间的复合物(Ras-Raf), 它是信号转导级联的中心. 这里是一个部分平衡的, 预先准备好的RAS-RAF复合物的pdb文件ras-raf.pdb.
结构中包含了ras和raf蛋白, 另外还有一个生理上必需的GTP核苷酸, 如下图所示:

出于本教程的目的, 为简单起见, 我们在计算中不处理GTP分子, 因为这需要为该化合物设置新的参数, 内容超出了本教程的范围. 因此, 我们从pdb文件中删除它, 这样就简单地将它从计算中删除了. 虽然并非严格正确, 但这种近似是合理的, 因为从上图可以看出, GTP并不直接参与结合面.
另外, 这个蛋白质中还含有一个镁离子, 它主要与GTP分子结合, 因此我们也将其删除. 因此, 你应该从pdb文件中删除残基243和244.
下一步是将res-raf.pdb文件分成两个独立的结构, 这样就有了ras-raf.pdb, ras.pdb和raf.pdb. 我们将使用这三个结构创建三个气相prmtop和inpcrd文件组用于MM-PBSA计算, 以及溶剂化复合物的一个文件组用于运行MD模拟:
tleap -s -f oldff/leaprc.ff14SB
注意: 对AMBER 14, tleap调用中请使用-f $AMBERHOME/dat/leap/cmd/oldff/leaprc.ff99来加载ff99力场
com = loadpdb ras-raf.pdb
ras = loadpdb ras.pdb
raf = loadpdb raf.pdb
确保为要使用的计算方法选择了正确的半径. 详细信息见手册中LEaP节的PBRadii部分. 如果需要设定特殊的原子半径, 可以参考推荐recommended_settings.pdf.
set default PBRadii mbondi2
saveamberparm com ras-raf.prmtop ras-raf.inpcrd
saveamberparm ras ras.prmtop ras.inpcrd
saveamberparm raf raf.prmtop raf.inpcrd
退出tleap前, 还要创建溶剂化的复合物用于MD模拟:
charge com
> Total unperturbed charge: -0.000000
> Total perturbed charge: -0.000000 (因此无需添加抗衡离子)
solvatebox com TIP3PBOX 12.0
saveamberparm com ras-raf_solvated.prmtop ras-raf_solvated.inpcrd
quit
上述命令的输入可以使用脚本进行简化:
tleap -s -f tleap.in > tleap.out
其中tleap.in文件内容为:
| tleap.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | source leaprc.protein.ff14SB
source leaprc.water.spce
source leaprc.gaff
com = loadpdb ras-raf.pdb
ras = loadpdb ras.pdb
raf = loadpdb raf.pdb
set default PBRadii mbondi2
saveamberparm com ras-raf.prmtop ras-raf.inpcrd
saveamberparm ras ras.prmtop ras.inpcrd
saveamberparm raf raf.prmtop raf.inpcrd
charge com
solvatebox com TIP3PBOX 12.0
saveamberparm com ras-raf_solvated.prmtop ras-raf_solvated.inpcrd
quit
|
下载相关文件:
ras-raf.prmtop,ras-raf.inpcrdras.prmtop,ras.inpcrdraf.prmtop,raf.inpcrdras-raf_solvated.prmtop,ras-raf_solvated.inpcrd
1.1 平衡溶剂化复合体
我们将对溶剂化复合物按以下步骤进行平衡: 短的能量最小化, 50 ps的升温, 对复合物进行弱限制条件下50 ps的密度平衡, 300 K下500 ps的等压平衡. 所有的模拟在运行时都对氢原子使用SHAKE约束, 并使用2 fs的时间步长和Langevin动力学控制温度. 输入文件如下:
| min.in | |
|---|---|
1 2 3 4 5 6 7 8 9 | minimise ras-raf
&cntrl
imin=1,maxcyc=1000,ncyc=500,
cut=8.0,ntb=1,
ntc=2,ntf=2,
ntpr=100,
ntr=1, restraintmask=':1-242',
restraint_wt=2.0
/
|
| heat.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | heat ras-raf
&cntrl
imin=0,irest=0,ntx=1,
nstlim=25000,dt=0.002,
ntc=2,ntf=2,
cut=8.0, ntb=1,
ntpr=500, ntwx=500,
ntt=3, gamma_ln=2.0,
tempi=0.0, temp0=300.0, ig=-1,
ntr=1, restraintmask=':1-242',
restraint_wt=2.0,
nmropt=1
/
&wt TYPE='TEMP0', istep1=0, istep2=25000,
value1=0.1, value2=300.0, /
&wt TYPE='END' /
|
| density.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 | heat ras-raf
&cntrl
imin=0,irest=1,ntx=5,
nstlim=25000,dt=0.002,
ntc=2,ntf=2,
cut=8.0, ntb=2, ntp=1, taup=1.0,
ntpr=500, ntwx=500,
ntt=3, gamma_ln=2.0,
temp0=300.0, ig=-1,
ntr=1, restraintmask=':1-242',
restraint_wt=2.0,
/
|
| equil.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 | heat ras-raf
&cntrl
imin=0,irest=1,ntx=5,
nstlim=250000,dt=0.002,
ntc=2,ntf=2,
cut=8.0, ntb=2, ntp=1, taup=2.0,
ntpr=1000, ntwx=1000,
ntt=3, gamma_ln=2.0,
temp0=300.0, ig=-1,
/
|
小心: 在本教程的示例中, 我们不会更改用于随机数生成器的随机种子的值, 随机种子是由命名列表变量ig控制的. 这主要是为了能够重复教程设置所得的结果. 但是, 在运行成品模拟时, 特别是使用ntt=2或3(Anderson或Langevin恒温器)时, 每次 重新启动MD时必须修改随机数种子的默认值. 如果您正在使用AMBER 10(的bugfix.26或更高版本)或AMBER 11或更高版本, 可以通过在cntrl命名列表中设置ig=-1自动执行此操作. 或者, 可以在每次重新开始计算时为ig指定一个你选择的正的随机数. 关于不这样做的误区的更多细节可参考文献: Cerutti DS, Duke, B., et al., “A Vulnerability in Popular Molecular Dynamics Packages Concerning Langevin and Andersen Dynamics”, JCTC, 2008, 4, 1669-1680
你可以使用下面的命令运行所有的4个模拟:
$AMBERHOME/bin/sander -O -i min.in -o min.out -p ras-raf_solvated.prmtop -c ras-raf_solvated.inpcrd -r min.rst -ref ras-raf_solvated.inpcrd
$AMBERHOME/bin/sander -O -i heat.in -o heat.out -p ras-raf_solvated.prmtop -c min.rst -r heat.rst -x heat.mdcrd -ref min.rst
gzip -9 heat.mdcrd
$AMBERHOME/bin/sander -O -i density.in -o density.out -p ras-raf_solvated.prmtop -c heat.rst -r density.rst -x density.mdcrd -ref heat.rst
gzip -9 density.mdcrd
$AMBERHOME/bin/sander -O -i equil.in -o equil.out -p ras-raf_solvated.prmtop -c density.rst -r equil.rst -x equil.mdcrd
gzip -9 equil.mdcrd
在1.7GHz, 16核的IBM P690机器上, 完成上面的模拟大于需要5小时.
可以在这里下载输出文件: equil.tar.gz
在我们进行MM-PBSA的成品模拟之前, 我们需要验证体系是否已经平衡. 为此, 我们从温度, 密度, 总能量和RMSD四个方面进行分析. 我们可以使用perl脚本(process_mdout.pl)从输出文件中提取有用的信息. 命令如下:
process_mdout.pl heat.out density.out equil.out
此外, 我们还要检查相对于最小化结构的蛋白质骨架算RMSD, 以确定在平衡期间构象是否已经稳定. 这可以使用measure_equil_rmsd.ptraj脚本借助ptraj或cpptraj完成:
| measure_equil_rmsd.ptraj | |
|---|---|
1 2 3 | trajin equil.mdcrd.gz 1 250 1
reference ras-raf_solvated.inpcrd
rms reference out equil.rmsd @CA,C,N
|
由于第一次对复合物体系的升温模拟是在等容条件下进行的, 并没有记录体系的密度数据. 因此需要编辑summary.DENSITY文件删除前50行(之所以这么做是因为xmgrace太愚蠢无法处理这种情况).
对复合物体系的温度, 密度, 总能量和RMSD进行绘图:
xmgrace summary.DENSITY
xmgrace summary.TEMP
xmgrace summary.ETOT
xmgrace equil.rmsd
结果如下:
- 密度

- 温度

- 总能量

- RMSD

在平衡期结束时, 密度, 温度和总能量曲线都明显地收敛. 看起来开始稳定的RMSD似乎没有完全收敛, 但鉴于本教程的目的也是可以接受的. 在实际计算中, 根据体系的大小, 我们需要运行更长的平衡时间. 接下来我们将运行成品模拟.
2. 运行成品模拟获得轨迹快照集
运行成品模拟时应该使用与平衡的最后阶段相同的条件, 以防止由于模拟条件变化而引起的势能突变.
我们将运行总共2 ns的成品模拟, 每10 ps记录一次坐标. 10 ps的时间间隔足够大能保证结构彼此是不相关的. 取决于你研究的体系, 使用更小时间间隔的快照也可能获得好的结果. 只要你获得的所有结构彼此不相关, 快照数目越多, 结果的统计误差应该越低. 对于RAS-RAF复合物这样的体系, 2 ns的模拟时间可能太短, 不足以获得足够多的不相关快照以对平衡系综进行充分采样. 20 ns左右的模拟时长可能更合适. 但是, 2 ns对于本教程来说已经足够了.
输入文件prod.in如下:
| prod.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 | prod ras-raf
&cntrl
imin=0,irest=1,ntx=5,
nstlim=250000,dt=0.002,
ntc=2,ntf=2,
cut=8.0, ntb=2, ntp=1, taup=2.0,
ntpr=5000, ntwx=5000,
ntt=3, gamma_ln=2.0,
temp0=300.0, ig=-1,
/
|
应该运行4次以获得2 ns的模拟时间. 由于这是一个简单的周期性边界的PME模拟, 如果需要可以使用PMEMD来进行模拟. PMEMD通常性能更好, 并且可以并行扩展. 下面是我在San Diego超算中心的Teragrid集群上使用96个核来运行上面的作业所用的PBS脚本run.x:
| run.x | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | #SDSC Teragrid PBS Script
#PBS -j oe
#PBS -l nodes=48:ppn=2
#PBS -l walltime=12:00:00
#PBS -q dque
#PBS -V
#PBS -M name@email.com
#PBS -A account_no
#PBS -N run_pmemd_96
cd /gpfs/projects/prod/
mpirun -v -machinefile $PBS_NODEFILE -np 96 /usr/local/apps/amber9/exe/pmemd -O -i prod.in -o prod1.out -p ras-raf_solvated.prmtop -c equil.rst -r prod1.rst -x prod1.mdcrd
mpirun -v -machinefile $PBS_NODEFILE -np 96 /usr/local/apps/amber9/exe/pmemd -O -i prod.in -o prod2.out -p ras-raf_solvated.prmtop -c prod1.rst -r prod2.rst -x prod2.mdcrd
mpirun -v -machinefile $PBS_NODEFILE -np 96 /usr/local/apps/amber9/exe/pmemd -O -i prod.in -o prod3.out -p ras-raf_solvated.prmtop -c prod2.rst -r prod3.rst -x prod3.mdcrd
mpirun -v -machinefile $PBS_NODEFILE -np 96 /usr/local/apps/amber9/exe/pmemd -O -i prod.in -o prod4.out -p ras-raf_solvated.prmtop -c prod3.rst -r prod4.rst -x prod4.mdcrd
gzip -9 prod*.mdcrd
|
下载输出文件: prod.tar.gz(84.8 MB)
为了获得好的结果, 体系在成品模拟阶段仍然在探索平衡相空间至关重要. 我们将通过绘制密度, 温度, 总能量和蛋白骨架RMSD图来检查是否如此, 所用方法与平衡阶段最后的检查步骤一样.
绘图结果如下:
- 密度

- 温度

- 总能量

- 骨架RMSD

从成品模拟RMSD的波动趋势上看, 体系还没有达到平衡状态, 但其他性质基本上是恒定的(注意纵轴标尺的大小). 理想情况下, 我们应该延长成品模拟的时长(约20 ns). 然而, 鉴于本教程的目的, 我们就使用2 ns的轨迹进行下面的步骤.
在下一节中我们将计算结合自由能, 可采用的方式有两种: 第一种使用(需要先安装)Python脚本MMPBSA.py, 第二种使用Perl脚本mm_pbsa.pl.
3. 计算结合自由能并分析结果
使用Perl脚本mm_pbsa.pl计算结合自由能
我们需要从成品模拟轨迹中抽取快照(不含溶剂水)用于MM-PBSA计算. mm_pbsa.pl脚本(位于$AMBERHOME/bin目录)可以自动完成这个提取过程. 请注意, 如果没有安装gzcat, 则需要在运行mm_pbsa之前将轨迹文件解压缩. 我们必须提供如下的输入文件extract_coords.mmpbsa(下载的文件中包含对每项的解释说明):
| extract_coords.mmpbsa | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 | @GENERAL
PREFIX snapshot
PATH ./
COMPLEX 1
RECEPTOR 1
LIGAND 1
COMPT ./ras-raf.prmtop
RECPT ./ras.prmtop
LIGPT ./raf.prmtop
GC 1
AS 0
DC 0
MM 0
GB 0
PB 0
MS 0
NM 0
@MAKECRD
BOX YES
NTOTAL 42193
NSTART 1
NSTOP 200
NFREQ 1
NUMBER_LIG_GROUPS 1
LSTART 2622
LSTOP 3862
NUMBER_REC_GROUPS 1
RSTART 1
RSTOP 2621
@TRAJECTORY
TRAJECTORY ./prod1.mdcrd
TRAJECTORY ./prod2.mdcrd
TRAJECTORY ./prod3.mdcrd
TRAJECTORY ./prod4.mdcrd
@PROGRAMS
|
该文件中指定了哪些原子属于受体, 配体和复合物, 并指定了对应于未溶剂化结构的prmtop文件, 轨迹中的快照总数, 提取步长和轨迹文件的名称. 我们还指定了每个输出文件都以snapshot作为前缀. 在本教程中, 我们定义RAS为受体, RAF为配体. 这单纯只是一个命名约定而已. 运行命令如下:
$AMBERHOME/bin/mm_pbsa.pl extract_coords.mmpbsa > extract_coords.log
此命令需要几分钟才能完成. 输出文件如下: extract_coords.tar.gz(14 MB)
如果发现任何错误, 可以检查日志文件, 同时确保盒子尺寸看起来合理. 如果发现盒子尺寸不合理, 可能是由于选择的原子数目有误或轨迹文件发生损坏.
基于已经提取出的快照, 我们可以计算复合物, 受体和配体的相互作用能和溶剂化自由能, 并对结果进行平均以获得结合自由能的估计值. 请注意, 在本教程中, 我们不会计算熵对结合能的贡献, 所以严格地说, 我们的结果并不是真正的自由能, 但可以用于比较相似的体系. 例如. 可以分析沿结合界面的氨基酸点突变的效果. 关于此的通常做法称为丙氨酸扫描.
作为演示, 我们将用MM_PBSA方法和MM_GBSA方法计算结合能, 这是通过mm_pbsa.pl的如下输入文件binding_energy.mmpbsa实现的(下载的文件中有注释):
| binding_energy.mmpbsa | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 | VERBOSE 0
PARALLEL 0
PREFIX snapshot
PATH ./
START 1
STOP 200
OFFSET 1
COMPLEX 1
RECEPTOR 1
LIGAND 1
COMPT ./ras-raf.prmtop
RECPT ./ras.prmtop
LIGPT ./raf.prmtop
GC 0
AS 0
DC 0
MM 1
GB 1
PB 1
MS 1
NM 0
@PB
PROC 2
REFE 0
INDI 1.0
EXDI 80.0
SCALE 2
LINIT 1000
ISTRNG 0.0
RADIOPT 0
ARCRES 0.0625
INP 1
SURFTEN 0.005
SURFOFF 0.00
IVCAP 0
CUTCAP -1.0
XCAP 0.0
YCAP 0.0
ZCAP 0.0
@MM
DIELC 1.0
@GB
IGB 2
GBSA 1
SALTCON 0.00
EXTDIEL 80.0
INTDIEL 1.0
SURFTEN 0.005
SURFOFF 0.00
@MS
PROBE 0.0
@PROGRAMS
|
该输入文件的各个部分指定了需要运行的计算, 计算时需要的文件以及计算结合自由能的不同贡献时所需要的所有特殊参数. 如果你打开下载的文件, 可以看到其中对不同项的解释说明.
不同参数的数值是根据经验数据确定的, 有待于当前研究的验证. 对于非极性溶剂化自由能的计算, Recommendation_setting.pdf文件给出当前的推荐设置. 计算结合自由能的示例输入文件以及常用的参数设置可以在$AMBERHOME/AmberTools/src/mm_pbsa/Examples/TEMPLATE_INPUT_SCRIPTS目录中找到. 更多信息请参考相关文献. 请注意, 早期版本的AMBER需要额外的Poisson-Boltzmann求解器, 如DelPhi, 但自AMBER 8起可以使用自带的PBSA程序. 这个程序计算速度有了明显提高, 并且更容易整合到AMBER中. 你可以使用如下命令运行:
$AMBERHOME/bin/mm_pbsa.pl binding_energy.mmpbsa > binding_energy.log
可以使用如下命令监控计算进度:
tail -f binding_energy.log
该计算大约需要2个小时才能完成(P4 3.2 GHz). 对600帧快照中的每一帧进行PBSA计算耗费了大部分时间. GBSA计算部分几秒钟内就能完成. 为了加快计算, 可以在PARALLEL下指定可用的处理器的数目, 这样mm_pbsa分析就可以并行, 能同时处理多个快照.
计算结束后, 会得到下列输出文件: binding_energy.log, snapshot_statistics.out, snapshot_com.all.out, snapshot_rec.all.out, snapshot_lig.all.out
binding_energy.log文件只显示了计算是否成功完成. all.out文件给出了每个物种每一快照的单独的能量贡献, 而statistics.out文件包含了平均结合自由能的最终结果:
| snapshot_statistics.out | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 | # COMPLEX RECEPTOR LIGAND
# ----------------------- ----------------------- -----------------------
# MEAN STD MEAN STD MEAN STD
# ======================= ======================= =======================
ELE -8656.78 70.18 -5602.09 63.10 -2102.25 52.57
VDW -984.99 24.34 -661.18 20.33 -256.02 12.93
INT 5085.33 50.22 3449.57 38.65 1635.76 29.42
GAS -4556.44 75.96 -2813.70 65.21 -722.52 53.50
PBSUR 65.09 1.05 45.25 0.64 27.24 0.46
PBCAL -3223.64 58.68 -2490.86 48.73 -1671.27 47.46
PBSOL -3158.55 58.26 -2445.62 48.45 -1644.03 47.31
PBELE -11880.42 34.25 -8092.96 29.34 -3773.52 17.30
PBTOT -7714.99 48.25 -5259.32 36.97 -2366.55 26.61
GBSUR 65.09 1.05 45.25 0.64 27.24 0.46
GB -3407.82 58.49 -2631.83 50.08 -1731.06 47.68
GBSOL -3342.74 58.15 -2586.58 49.83 -1703.82 47.55
GBELE -12064.60 26.94 -8233.92 23.57 -3833.31 13.40
GBTOT -7899.17 47.07 -5400.28 35.65 -2426.34 26.80
# DELTA
# -----------------------
# MEAN STD
# =======================
ELE -952.43 44.10
VDW -67.79 5.18
INT -0.00 0.00
GAS -1020.22 44.58
PBSUR -7.40 0.41
PBCAL 938.50 42.51
PBSOL 931.09 42.31
PBELE -13.94 9.43
PBTOT -89.13 7.94
GBSUR -7.40 0.41
GB 955.07 41.30
GBSOL 947.66 41.10
GBELE 2.63 7.41
GBTOT -72.56 6.40
|
该文件中不同项的含义如下:
ELE: 由分子力场(MM)计算的静电能VDW: 来自MM的范德华贡献INT: MM力场中由键, 角和二面角项引起的内能(在单轨迹方法中该项总是等于零)GAS: 总的气相能量(ELE,VDW和INT的总和)PBSUR/GBSUR: 由经验模型计算的溶剂化自由能中的非极性贡献PBCAL/GB: 分别由PB或GB计算的溶剂化自由能中的静电贡献PBSOL/GBSOL: 溶剂化的非极性和极性贡献之和PBELE/GBELE: 静电溶剂化自由能和MM静电能的总和PBTOT/GBTOT: 根据上述项得出的最终结合自由能的估计(kcal/mol)
值得注意的是, 通常情况下, 结合自由能的主要贡献来自ELE, PBCAL/GBCAL和VDW部分, 前两项大致相互抵消, 这可以通过查看PBELE/GBELE的值是否远小于ELE或PBCAL/GBCAL对它的贡献进行检查.
通常我们会期望能发现非常有利的静电能和不利的溶剂化自由能, 这意味着结合粒子去溶剂化并对齐到结合界面的能量.
总结合自由能-89.13 kcal/mol为负值, 这表明在纯水中, 该蛋白-蛋白复合体的结合是有利的. 但是, 要记住, 该结果并不等于真正的结合自由能, 因为我们没有考虑(不利的)熵对结合自由能的贡献. 注意到, GB方法给出的结合能稍低, 但仍然表明这是一个有利的结合状态.
若要扩展本教程, 可以研究更改溶剂的盐含量, 修改特定残基或选择不同的起始结构对于结合自由能的影响. 另外, 也可以考虑利用nmode来计算熵变, 但要注意, 对于这种规模的复合物来说, 计算熵将会耗费大量的内存和时间.
利用Python脚本MMPBSA.py计算结合自由能

在本教程中, 我们将演示使用AmberTools中发布的MM-PBSA方法计算结合自由能, 运行丙氨酸扫描, 进行简正模式分析以计算熵变. 教程分为如下几部分:
3.1 计算蛋白-蛋白复合物(Ras-Raf)的结合自由能
本小节中, 我们将模拟人类H-Ras蛋白与结合于Ras结合结构域的C-Raf1形成的复合物(Ras-Raf), 该复合物是信号转导级联的中心. 部分平衡的经过预处理的RAS-RAF复合体的pdb结构如下图所示. 该结构中含有ras和raf蛋白, 另外还有一个生理必需的GTP核酸.
关于如何构建初始结构和运行动力学模拟以获得平衡体系, 请参考前面几节.
使用MMPBSA.py计算结合自由能的重要文件是拓扑文件和mdcrd文件(ras-raf_top_mdcrd.tgz).
我们现在将计算复合物, 受体和配体的相互作用能和溶剂化自由能, 并对结果进行平均以获得结合自由能的估计值. 请注意, 在教程的这一部分我们不计算熵对结合能的贡献, 因此严格来说, 所得的结果并不是真正的自由能, 但可以用来对相似的体系进行比较. 有关使用简正模式分析(Nmode)计算体系的熵贡献, 可参考3.5节. 也可以取消下面输入文件中&general命名列表中最后一行的注释, 以便使用AMBER的ptraj模块进行准简谐熵计算.
我们将分别使用MM-GBSA方法和MM-PBSA方法进行结合自由能的计算并进行比较. 以下为MMPBSA.py的输入文件mmpbsa.in:
| mmpbsa.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 | Input file for running PB and GB
&general
endframe=50, verbose=1,
# entropy=1,
/
&gb
igb=2, saltcon=0.100
/
&pb
istrng=0.100,
/
|
MMPBSA.py的输入文件与AMBER的sander模块所用的mdin输入文件类似. 每个命名列表都是以&符号开始, 后面跟着命名列表的名称. 另外, 反斜线(/)或&end可用于结束命名列表. 有关所有变量的完整列表, 请参阅AMBER手册或在终端输入$AMBERHOME/bin/MMPBSA.py --input-file-help. mmpbsa.in被分成三个命名列表: general, pb和gb. general命名列表旨在指定并非仅适用于计算的特定部分, 而是针对所有部分的变量. 在本教程中, 我们将RAS定义为受体, RAF定义为配体. endframe变量设置轨迹mdcrd中要停止的帧. &gb和&pb命名列表中给出了用于MM-GBSA和MM-PBSA计算的参数值. verbose变量用于指定输出文件的详细程度.
使用如下命令运行文件:
$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp ras-raf_solvated.prmtop -cp ras-raf.prmtop -rp ras.prmtop -lp raf.prmtop -y *.mdcrd
这将交互式地运行脚本, 并将计算进度输出至标准输出终端, 将任何错误或警告输出至标注错误终端. 最后, 计算完成后会在终端上输出总耗时以及计算过程中每一步骤的耗时.
另外, 命令行参数可以用shell识别的通配符(即bash的*和?). 例如, 命令行中的-y *.mdcrd会告知程序读入工作目录中所有以.mdcrd结尾的文件, 并将其作为待分析的轨迹.
下载脚本生成的输出文件: `pb_gb_output1.tgz.
脚本使用ptraj生成了三个非溶剂化的mdcrd文件(复合物, 受体, 配体), 它们是在GB和PB计算过程中分析过的坐标. *.mdout文件包含所有指定帧的能量.
还会生成一个平均结构的PDB文件, 其中的结构(通过RMS)对齐到所有快照. 如果需要, 可以使用ptraj对这个结构进行准简谐熵计算. MMPBSA.py生成的所有文件的名称均以前缀_MMPBSA_开始, 除了最终的输出文件FINAL_RESULTS_MMPBSA.dat之外:
| FINAL_RESULTS_MMPBSA.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 | | Run on Thu Feb 11 12:18:37 EST 2010
|Input file:
|--------------------------------------------------------------
|Input file for running PB and GB
|&general
| endframe=50, verbose=1,
|# entropy=1,
|/
|&gb
| igb=2, saltcon=0.100
|/
|&pb
| istrng=0.100,
|/
|--------------------------------------------------------------
|Solvated complex topology file: ras-raf_solvated.prmtop
|Complex topology file: ras-raf.prmtop
|Receptor topology file: ras.prmtop
|Ligand topology file: raf.prmtop
|Initial mdcrd(s): prod.mdcrd
|
|Best guess for receptor mask: ":1-166"
|Best guess for ligand mask: ":167-242"
|Calculations performed using 50 frames.
|Poisson Boltzmann calculations performed using internal PBSA solver in sander.
|
|All units are reported in kcal/mole.
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
GENERALIZED BORN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1863.7944 17.1704 2.4283
EEL -17200.7297 75.9366 10.7391
EGB -3249.6511 65.2075 9.2217
ESURF 91.3565 1.3938 0.1971
G gas -19064.5240 77.8536 11.0102
G solv -3158.2946 65.2224 9.2238
TOTAL -22222.8186 51.0216 7.2155
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1268.1888 14.2342 2.0130
EEL -11557.0773 71.7127 10.1417
EGB -2532.0669 57.7003 8.1600
ESURF 64.2843 1.1143 0.1576
G gas -12825.2661 73.1118 10.3396
G solv -2467.7826 57.7110 8.1616
TOTAL -15293.0487 35.3527 4.9996
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.4198 1.3322
EEL -4684.4720 36.1449 5.1117
EGB -1688.9631 26.5353 3.7527
ESURF 37.0493 0.6185 0.0875
G gas -5213.7811 37.3522 5.2824
G solv -1651.9138 26.5425 3.7537
TOTAL -6865.6949 25.8878 3.6611
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -66.2966 4.2751 0.6046
EEL -959.1803 34.9190 4.9383
EGB 971.3789 33.0497 4.6739
ESURF -9.9770 0.3759 0.0532
DELTA G gas -1025.4769 35.1797 4.9752
DELTA G solv 961.4018 33.0518 4.6742
DELTA G binding = -64.0750 +/- 6.3729 0.9013
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
POISSON BOLTZMANN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1863.7944 17.1704 2.4283
EEL -17200.7297 75.9366 10.7391
EPB -3207.7160 66.4023 9.3907
ECAVITY 67.8762 0.7818 0.1106
G gas -19064.5240 6061.1875 857.1813
G solv -3139.8399 66.4069 9.3914
TOTAL -7686.8660 52.5400 7.4303
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1268.1888 14.2342 2.0130
EEL -11557.0773 71.7127 10.1417
EPB -2483.7242 56.4551 7.9840
ECAVITY 47.1495 0.4737 0.0670
G gas -12825.2661 5345.3320 755.9441
G solv -2436.5747 56.4571 7.9842
TOTAL -5250.2060 38.5188 5.4474
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.4198 1.3322
EEL -4684.4720 36.1449 5.1117
EPB -1670.4169 27.6694 3.9131
ECAVITY 28.0328 0.4133 0.0584
G gas -5213.7811 1395.1865 197.3092
G solv -1642.3841 27.6725 3.9135
TOTAL -2350.3020 25.1197 3.5525
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -66.2966 4.2751 0.6046
EEL -959.1803 34.9190 4.9383
EPB 946.4251 34.5128 4.8808
ECAVITY -7.3062 0.3004 0.0425
DELTA G gas -1025.4769 1237.6138 175.0250
DELTA G solv 939.1189 34.5141 4.8810
DELTA G binding = -86.3579 +/- 8.3264 1.1775
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
WARNINGS:
igb=2 should be used with mbondi2 pbradii set. Yours are modified Bondi radii (mbondi)
|
统计文件的开始部分包括日期/时间, 基于给定值和文件的任何警告, mmpbsa.in输入文件内容, 脚本所用的文件的名称, 分析的帧数以及使用了哪个PB求解器(如果使用的话). 统计文件的其余内容包括所有的平均能量, 标准偏差和平均值的标准误差, 先列出GB对应的值, 再列出PB对应的值. 每个部分之后, 会给出结合自由能ΔG及误差. 文件中不同项的含义如下:
VDWAALS: 来自分子力场(MM)的范德华贡献EEL: 由MM力场计算的静电能EPB/EGB: 分别由PB或GB计算的静电对溶剂化自由能的贡献ECAVITY: 通过经验模型计算的非极性对溶剂化自由能的贡献DELTA G binding: 根据上面各项计算出的最终结合自由能的估计值(kcal/mol)
值得注意的是, 结果中并未报告总的气相能量, 因为使用单一轨迹计算方法时, 受体和配体的成键势能项的值会精确地与复合体中的对应值互相抵消. 如果这两项能量在允许的误差范围内不能抵消, 将会给出错误信息.
通常我们会期望能发现非常有利的静电能和不利的溶剂化自由能, 这意味着结合粒子去溶剂化并对齐到结合界面的能量.
总结合自由能-86.36 kcal/mol为负值, 这表明在纯水中, 该蛋白-蛋白复合体的结合是有利的. 但是, 要记住, 该结果并不等于真正的结合自由能, 因为我们没有考虑(不利的)熵对结合自由能的贡献. 注意到, GB方法给出的结合能稍低, 但仍然表明这是一个有利的结合状态.
3.2 使用三个处理器并行计算Ras-Raf的结合自由能
在本节中, 我们将并行地计算结合自由能. MMPBSA.py.MPI通过为每个线程(进程)分配相同数目的帧进行并行化计算. 因此, 当处理的帧数是所启动的线程数的倍数时, 它的运行效率最高. 但是, 这并不是必须的. 如果帧数不是线程数的倍数, 则”剩余”帧将在启动线程数的一个子集中均匀分配. 例如, 在3个线程上计算50帧将导致2个线程计算17帧, 最后一个线程只计算16帧. 因此, 第三个线程将不得不等待前两个线程完成计算后才能继续进行下面的计算. 出于这个原因, 使用5个线程将是更明智的选择(每个线程处理10帧). 但是, 线程数不能超过要处理的帧数, 否则MMPBSA.py.MPI将会终止, 给出错误信息. MMPBSA.py.MPI的输入文件与MMPBSA.py的输入文件完全相同:
| mmpbsa.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 | Input file for running PB and GB
&general
endframe=50, verbose=1,
# entropy=1,
/
&gb
igb=2, saltcon=0.100
/
&pb
istrng=0.100,
/
|
运行命令如下:
mpirun -np 4 $AMBERHOME/bin/MMPBSA.py.MPI -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp ras-raf_solvated.prmtop -cp ras-raf.prmtop -rp ras.prmtop -lp raf.prmtop -y *.mdcrd > progress.log 2>&1
或者使用下面的命令将作业脚本提交到排队系统, 如PBS系统:
qsub parallel.job
作业脚本parallel.job内容如下:
| parallel.job | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 | #!/bin/sh
#PBS -N rasraf_parallel
#PBS -o parallel.out
#PBS -e parallel.err
#PBS -m abe
#PBS -M email@address.com
#PBS -q brute
#PBS -l nodes=1:node:ppn=4
#PBS -l pmem=900mb
cd $PBS_O_WORKDIR
mpirun -np 4 MMPBSA.py.MPI -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp ras-raf_solvated.prmtop -cp ras-raf.prmtop -rp ras.prmtop -lp raf.prmtop -y bigprod.mdcrd > progress.log 2>&1
|
计算进度会输出到文件progress.log. 计算过程中的所有错误也会输出到这个文件中(这是2>&1的目的). 最后, 计算完成后会显示每个步骤所用的时间.
| progress.log | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 | MMPBSA.py.MPI being run on 4 processors
ptraj found! Using /scr/arwen_3/swails/i686/amber11/exe/ptraj
sander found! Using /scr/arwen_3/swails/i686/amber11/exe/sander
Preparing trajectories with ptraj...
50 frames were read in and processed by ptraj for use in calculation.
Starting calculations
Starting gb calculation...
Starting pb calculation...
Calculations complete. Writing output file(s)...
Timing:
Processing Trajectories With Ptraj: 0.126 min.
Total GB Calculation Time (sander): 4.782 min.
Total PB Calculation Time (sander): 28.407 min.
Output File Writing Time: 0.053 min.
Total Time Taken: 33.379 min.
MMPBSA Finished. Thank you for using. Please send any bugs/suggestions/comments to mmpbsa.amber@gmail.com
|
keep_files设置为默认值1, 输出文件为Parallel_output.tgz
最终结果为FINAL_RESULTS_MMPBSA.dat:
| FINAL_RESULTS_MMPBSA.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 | | Run on Sun Feb 14 19:10:43 EST 2010
|Input file:
|--------------------------------------------------------------
|Input file for running PB and GB in serial
|&general
| endframe=50, verbose=1,
| mpi_cmd='mpirun -np 3', nproc=3
|/
|&gb
| igb=2, saltcon=0.100
|/
|&pb
| istrng=0.100,
|/
|--------------------------------------------------------------
|Solvated complex topology file: ras-raf_solvated.prmtop
|Complex topology file: ras-raf.prmtop
|Receptor topology file: ras.prmtop
|Ligand topology file: raf.prmtop
|Initial mdcrd(s): bigprod.mdcrd
|
|Best guess for receptor mask: ":1-166"
|Best guess for ligand mask: ":167-242"
|Calculations performed using 50 frames.
|Poisson Boltzmann calculations performed using internal PBSA solver in sander.
|
|All units are reported in kcal/mole.
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
GENERALIZED BORN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1863.7944 17.1704 2.4283
EEL -17200.7297 75.9366 10.7391
EGB -3249.6511 65.2075 9.2217
ESURF 91.3565 1.3938 0.1971
G gas -19064.5240 77.8536 11.0102
G solv -3158.2946 65.2224 9.2238
TOTAL -22222.8186 51.0216 7.2155
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1268.1888 14.2342 2.0130
EEL -11557.0773 71.7127 10.1417
EGB -2532.0669 57.7003 8.1600
ESURF 64.2843 1.1143 0.1576
G gas -12825.2661 73.1118 10.3396
G solv -2467.7826 57.7110 8.1616
TOTAL -15293.0487 35.3527 4.9996
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.4198 1.3322
EEL -4684.4720 36.1449 5.1117
EGB -1688.9631 26.5353 3.7527
ESURF 37.0493 0.6185 0.0875
G gas -5213.7811 37.3522 5.2824
G solv -1651.9138 26.5425 3.7537
TOTAL -6865.6949 25.8878 3.6611
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -66.2966 4.2751 0.6046
EEL -959.1803 34.9190 4.9383
EGB 971.3789 33.0497 4.6739
ESURF -9.9770 0.3759 0.0532
DELTA G gas -1025.4769 35.1797 4.9752
DELTA G solv 961.4018 33.0518 4.6742
DELTA G binding = -64.0750 +/- 6.3729 0.9013
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
POISSON BOLTZMANN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1863.7944 17.1704 2.4283
EEL -17200.7297 75.9366 10.7391
EPB -3207.7160 66.4023 9.3907
ECAVITY 67.8762 0.7818 0.1106
G gas -19064.5240 6061.1875 857.1813
G solv -3139.8399 66.4069 9.3914
TOTAL -7686.8660 52.5400 7.4303
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1268.1888 14.2342 2.0130
EEL -11557.0773 71.7127 10.1417
EPB -2483.7242 56.4551 7.9840
ECAVITY 47.1495 0.4737 0.0670
G gas -12825.2661 5345.3320 755.9441
G solv -2436.5747 56.4571 7.9842
TOTAL -5250.2060 38.5188 5.4474
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.4198 1.3322
EEL -4684.4720 36.1449 5.1117
EPB -1670.4169 27.6694 3.9131
ECAVITY 28.0328 0.4133 0.0584
G gas -5213.7811 1395.1865 197.3092
G solv -1642.3841 27.6725 3.9135
TOTAL -2350.3020 25.1197 3.5525
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -66.2966 4.2751 0.6046
EEL -959.1803 34.9190 4.9383
EPB 946.4251 34.5128 4.8808
ECAVITY -7.3062 0.3004 0.0425
DELTA G gas -1025.4769 1237.6138 175.0250
DELTA G solv 939.1189 34.5141 4.8810
DELTA G binding = -86.3579 +/- 8.3264 1.1775
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
WARNINGS:
igb=2 should be used with mbondi2 pbradii set. Yours are modified Bondi radii (mbondi)
|
3.3: 计算蛋白-配体复合物(雌激素受体和雷洛昔芬)的结合自由能
1. 建立起始结构, 通过模拟获得平衡体系
本小节中, 我们将模拟雌激素受体蛋白和雷洛昔芬配体形成的蛋白-配体复合物. 其预处理过的pdb文件如下Estrogen_Receptor-Raloxifene.pdb.
该结构包含雌激素受体蛋白以及配体雷洛昔芬, 雷洛昔芬已经锚定在了受体蛋白上, 如下图所示:
这个体系的构建方式与第1节中的Ras-Raf体系类似. 关于如何构建起始结构和运行动力学模拟以获得平衡体系, 请参考第1节和第2节. 另外, 必须利用antechamber获得雷洛昔芬的正确参数. 详细说明请参考AMBER基础教程B4.
MD模拟中, 使用MMPBSA.py计算结合自由能所需的重要文件是MD模拟所用的拓扑文件和mdcrd文件, 下载Est_Rec_top_mdcrd.tgz.
2. 计算雌激素受体和雷洛昔芬的结合自由能
此节说明文件与前几节几乎相同, 故此省略, 只给出命令和结果.
MMPBSA.py计算的输入文件mmpbsa.in:
| mmpbsa.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 | Input file for running PB and GB
&general
endframe=50, keep_files=2,
/
&gb
igb=2, saltcon=0.100,
/
&pb
istrng=0.100,
/
|
运行命令:
$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp 1err.solvated.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y *.mdcrd
使用keep_files=2, 得到的输出文件为pb_gb_output2.tgz
最终结果
| FINAL_RESULTS_MMPBSA.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 | | Run on Thu Feb 11 12:44:26 EST 2010
|Input file:
|--------------------------------------------------------------
|Input file for running PB and GB
|&general
| endframe=50, keep_files=2,
|/
|&gb
| igb=2, saltcon=0.100,
|/
|&pb
| istrng=0.100,
|/
|--------------------------------------------------------------
|Solvated complex topology file: 1err.solvated.prmtop
|Complex topology file: complex.prmtop
|Receptor topology file: receptor.prmtop
|Ligand topology file: ligand.prmtop
|Initial mdcrd(s): 1err_prod.mdcrd
|
|Best guess for receptor mask: ":1-240"
|Best guess for ligand mask: ":241"
|Ligand residue name is "RAL"
|
|Calculations performed using 50 frames.
|Poisson Boltzmann calculations performed using internal PBSA solver in sander.
|
|All units are reported in kcal/mole.
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
GENERALIZED BORN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -2013.3801 20.3021 2.8712
EEL -16938.6450 85.7631 12.1287
EGB -3507.0086 67.7839 9.5861
ESURF 97.5448 1.3301 0.1881
G gas -18952.0251 88.1333 12.4639
G solv -3409.4639 67.7969 9.5879
TOTAL -22361.4889 47.1982 6.6748
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1955.2272 19.2311 2.7197
EEL -16895.0354 85.5797 12.1028
EGB -3528.7276 68.3585 9.6673
ESURF 101.2613 1.3071 0.1849
G gas -18850.2626 87.7138 12.4046
G solv -3427.4663 68.3710 9.6691
TOTAL -22277.7288 48.1057 6.8032
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1.8595 2.0516 0.2901
EEL -5.5796 2.0333 0.2876
EGB -28.4863 0.6040 0.0854
ESURF 4.4326 0.0462 0.0065
G gas -7.4391 2.8885 0.4085
G solv -24.0538 0.6058 0.0857
TOTAL -31.4929 5.0748 0.7177
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -56.2934 2.9265 0.4139
EEL -38.0300 3.2114 0.4542
EGB 50.2053 2.5869 0.3658
ESURF -8.1491 0.2589 0.0366
DELTA G gas -94.3234 4.3449 0.6145
DELTA G solv 42.0562 2.5999 0.3677
DELTA G binding = -52.2672 +/- 2.4568 0.3475
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
POISSON BOLTZMANN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -2013.3801 20.3021 2.8712
EEL -16938.6450 85.7631 12.1287
EPB -3329.1708 67.0354 9.4802
ECAVITY 68.2656 0.5195 0.0735
G gas -18952.0251 7767.4837 1098.4881
G solv -3260.9052 67.0374 9.4805
TOTAL -5265.0831 49.0426 6.9357
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1955.2272 19.2311 2.7197
EEL -16895.0354 85.5797 12.1028
EPB -3355.4746 67.3299 9.5219
ECAVITY 70.1184 0.5285 0.0747
G gas -18850.2626 7693.7163 1088.0558
G solv -3285.3562 67.3320 9.5222
TOTAL -5279.4509 50.4067 7.1286
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1.8595 2.0516 0.2901
EEL -5.5796 2.0333 0.2876
EPB -31.3364 0.6953 0.0983
ECAVITY 3.1896 0.0288 0.0041
G gas -7.4391 8.3434 1.1799
G solv -28.1468 0.6959 0.0984
TOTAL 56.0934 5.0476 0.7138
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -56.2934 2.9265 0.4139
EEL -38.0300 3.2114 0.4542
EPB 57.6402 3.0642 0.4333
ECAVITY -5.0423 0.1683 0.0238
DELTA G gas -94.3234 18.8778 2.6697
DELTA G solv 52.5978 3.0688 0.4340
DELTA G binding = -41.7256 +/- 2.9618 0.4189
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
|
3.4 计算Ras-Raf的结合自由能, 并利用丙氨酸扫描(Alanine Scanning)比较分析Ras-Raf复合物中某个残基突变为丙氨酸后的结果
1. 设置pdb文件, 为后续的tleap做准备
部分平衡后预处理过的RAS-RAF复合物的结构文件ras-raf.pdb
接下来我们必须准备突变体结构的pdb文件供tleap使用. 为了保证prmtop文件的一致性, 我们强烈建议在运行任何模拟之前准备该突变体的pdb及其拓扑文件, 并与第1节中创建的初始拓扑文件一起. 本教程中, 我们选择将残基21(异亮氨酸, I21)突变为丙氨酸, 因为I21位于受体和配体结合的界面处, 应该对结合能有明显的影响. 请注意, 本教程涉及的代码目前只适用于丙氨酸突变.
由于I21仅处于受体中, 所以我们不需要准备突变配体的pdb文件. 因此, 我们只需要修改ras-raf.pdb和ras.pdb. 为此, 你需要了解一些所涉及到的氨基酸结构的知识. 异亮氨酸的侧链是-CH(CH3)CH2CH3, 而丙氨酸的侧链是-CH3. 由于异亮氨酸侧链的原子数比丙氨酸侧链的多, 因此我们必须从pdb文件中去除原子及其相应的信息(名称, 编号, 坐标等). 这种突变涉及除β-C(CB)之外的所有侧链原子. 在I21中, 这意味着我们必须删除Ras-Raf和Ras的pdb文件中与原子294到305相对应的行. 我们不需要添加β-H(HB)原子, 因为基于为体系选择的特定库文件, tleap会将这些原子添加到合适的位置. 最后, 将残基21所有剩余的原子对应的残基名称从ILE更改为ALA. 此过程将生成两个突变的pdb文件: ras-raf_mutant.pdb和ras_mutant.pdb. RAS-RAF及其I21A突变结构如下图所示:
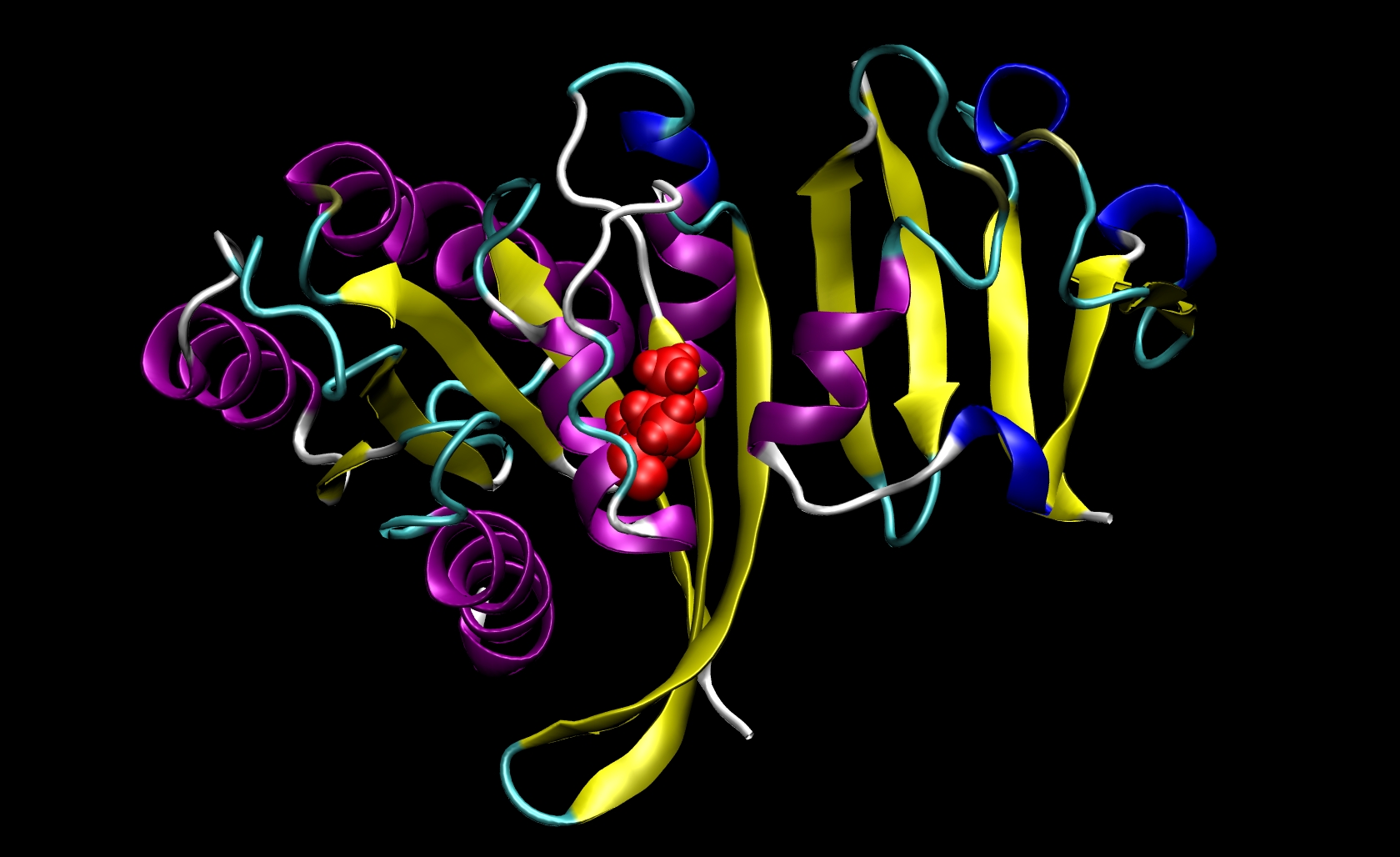
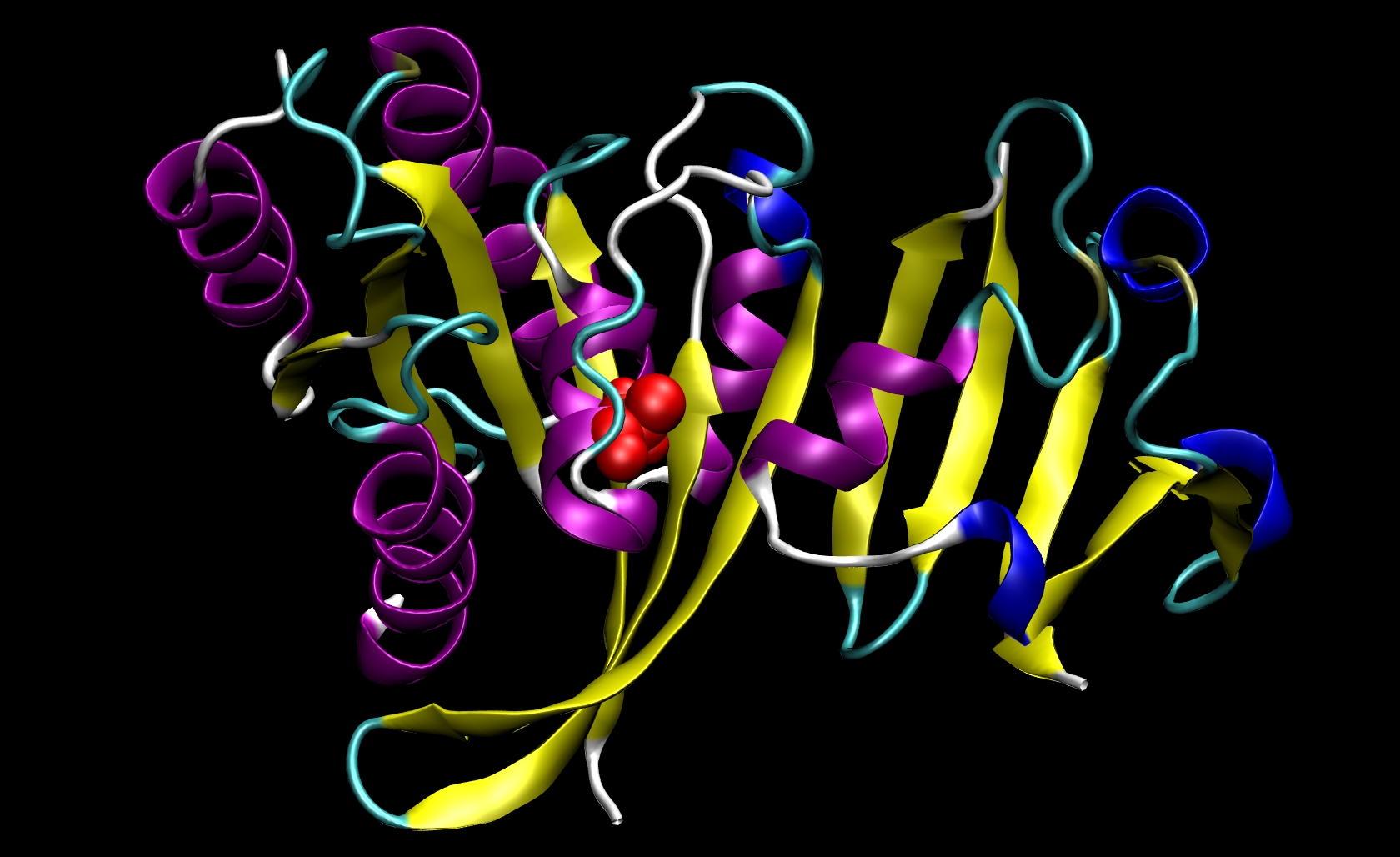
其他残基的突变也可以使用类似的方法, 其中位于CB之后羰基碳(C)之前的原子组可以从pdb文件中去除. 值得注意的是, 单次计算中只能进行一次突变.
2. 构建起始拓扑和坐标文件, 模拟获得平衡体系
基于已经生成的突变pdb文件, 可用tleap为这些结构构建相应的拓扑和坐标文件. 首先, 我们将生成对应非突变复合体的文件:
$AMBERHOME/exe/tleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99SB
com = loadpdb ras-raf.pdb
ras = loadpdb ras.pdb
raf = loadpdb raf.pdb
saveamberparm com ras-raf.prmtop ras-raf.inpcrd
saveamberparm ras ras.prmtop ras.inpcrd
saveamberparm raf raf.prmtop raf.inpcrd
构建用于MD模拟的溶剂化复合物:
charge com
> Total unperturbed charge: -0.000000
> Total perturbed charge: -0.000000 (Hence there is no need to add counter ions)
solvatebox com TIP3PBOX 12.0
saveamberparm com ras-raf_solvated.prmtop ras-raf_solvated.inpcrd
在退出tleap之前, 构建与突变pdb文件相应的拓扑和坐标文件:
com_mut = loadpdb ras-raf_mutant.pdb
ras_mut = loadpdb ras_mutant.pdb
saveamberparm com_mut rasraf_mutant.prmtop rasraf_mutant.inpcrd
saveamberparm ras_mut ras_mutant.prmtop ras_mutant.inpcrd
quit
上述步骤共生成了12个文件(6个.prmtop文件和6个.inpcrd文件). 非突变体.prmtop和.inpcrd文件用于运行MD模拟以获得平衡体系, 具体方法可参考第1节和第2节.
利用MMPBSA.py计算结合自由能的重要文件是(非突变体和突变体的)拓扑文件, 以及使用非突变体的拓扑文件和坐标文件运行得到的mdcrd文件ras-raf_alascan.tgz
3. 对Ras-Raf的结合自由能进行丙氨酸扫描计算
我们现在将计算复合物, 受体和配体的相互作用能和溶剂化自由能, 并对结果进行平均以获得结合自由能的估计值. 然后, 为了与”野生型”进行比较, 我们会对突变后的结构进行相同的计算, 计算前需要先将mdcrd中的坐标突变. 请注意, 在教程的这一部分我们不计算熵对结合能的贡献, 因此严格来说, 所得的结果并不是真正的自由能, 但可以用来对相似的体系进行比较. 有关使用简正模式分析(Nmode)计算体系的熵贡献, 可参考3.5节. 也可以取消下面输入文件中&general命名列表中最后一行的注释, 以便使用AMBER的ptraj模块进行准简谐熵计算.
计算方法与前面的相同, MMPBSA.py的输入文件mmpbsa.in如下:
| mmpbsa.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 | sample input file for running alanine scanning
&general
startframe=1, endframe=50, interval=1,
verbose=1,
/
&gb
saltcon=0.1
/
&pb
istrng=0.100
/
&alanine_scanning
/
|
文件中的&alanine_scanning命名列表表示初始化脚本中的丙氨酸扫描, 其中唯一可使用的变量是mutant_only, 其详细说明见手册.
运行命令如下:
$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -sp ras-raf_solvated.prmtop -cp rasraf.prmtop -rp ras.prmtop -lp raf.prmtop -y *.mdcrd -mc rasraf_mutant.prmtop -mr ras_mutant.prmtop
| FINAL_RESULTS_MMPBSA.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 | | Run on Thu Feb 11 13:11:48 EST 2010
|Input file:
|--------------------------------------------------------------
|sample input file for running alanine scanning
| &general
| startframe=1, endframe=50, interval=1,
| verbose=1,
|/
|&gb
| saltcon=0.1
|/
|&pb
| istrng=0.100
|/
|&alanine_scanning
|/
|--------------------------------------------------------------
|Solvated complex topology file: ras-raf_solvated.prmtop
|Complex topology file: rasraf.prmtop
|Receptor topology file: ras.prmtop
|Ligand topology file: raf.prmtop
|Initial mdcrd(s): bigprod.mdcrd
|Mutant complex topology file: rasraf_mutant.prmtop
|Mutant receptor topology file: ras_mutant.prmtop
|Mutant ligand topology file: raf.prmtop
|
|Best guess for receptor mask: ":1-166"
|Best guess for ligand mask: ":167-242"
|Calculations performed using 50 frames.
|Poisson Boltzmann calculations performed using internal PBSA solver in sander.
|
|All units are reported in kcal/mole.
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
GENERALIZED BORN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1863.7944 17.1704 2.4283
EEL -17200.7297 75.9366 10.7391
EGB -3142.2247 63.1977 8.9375
ESURF 91.3565 1.3938 0.1971
G gas -19064.5240 77.8536 11.0102
G solv -3050.8682 63.2131 8.9397
TOTAL -22115.3922 51.5332 7.2879
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1268.1888 14.2342 2.0130
EEL -11557.0773 71.7127 10.1417
EGB -2444.8629 54.9156 7.7662
ESURF 64.2843 1.1143 0.1576
G gas -12825.2661 73.1118 10.3396
G solv -2380.5786 54.9269 7.7678
TOTAL -15205.8447 36.8422 5.2103
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.4198 1.3322
EEL -4684.4720 36.1449 5.1117
EGB -1661.8286 26.5442 3.7539
ESURF 37.0493 0.6185 0.0875
G gas -5213.7811 37.3522 5.2824
G solv -1624.7794 26.5514 3.7549
TOTAL -6838.5604 25.6515 3.6277
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -66.2966 4.2751 0.6046
EEL -959.1803 34.9190 4.9383
EGB 964.4668 32.9201 4.6556
ESURF -9.9770 0.3759 0.0532
DELTA G gas -1025.4769 35.1797 4.9752
DELTA G solv 954.4898 32.9223 4.6559
DELTA G binding = -70.9871 +/- 6.6875 0.9457
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
I21A MUTANT:
GENERALIZED BORN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1855.4226 17.0765 2.4150
EEL -17210.2882 75.8866 10.7320
EGB -3145.1010 63.2477 8.9446
ESURF 91.8639 1.3913 0.1968
G gas -19065.7108 77.7842 11.0003
G solv -3053.2370 63.2630 8.9467
TOTAL -22118.9478 50.9582 7.2066
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1261.9126 14.1817 2.0056
EEL -11566.4419 71.5475 10.1183
EGB -2447.4831 55.0008 7.7783
ESURF 64.5090 1.1105 0.1570
G gas -12828.3545 72.9394 10.3152
G solv -2382.9741 55.0120 7.7799
TOTAL -15211.3287 36.2055 5.1202
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.4198 1.3322
EEL -4684.4720 36.1449 5.1117
EGB -1661.8286 26.5442 3.7539
ESURF 37.0493 0.6185 0.0875
G gas -5213.7811 37.3522 5.2824
G solv -1624.7794 26.5514 3.7549
TOTAL -6838.5604 25.6515 3.6277
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -64.2010 4.0841 0.5776
EEL -959.3742 34.9114 4.9372
EGB 964.2108 32.9092 4.6541
ESURF -9.6943 0.3800 0.0537
DELTA G gas -1023.5752 35.1495 4.9709
DELTA G solv 954.5164 32.9114 4.6544
DELTA G binding = -69.0588 +/- 6.5302 0.9235
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
RESULT OF ALANINE SCANNING: (I21A MUTANT:) DELTA DELTA G binding = 1.9283 +/- 9.3470
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
POISSON BOLTZMANN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1863.7944 17.1704 2.4283
EEL -17200.7297 75.9366 10.7391
EPB -3227.2145 64.4523 9.1149
ECAVITY 68.4754 0.7567 0.1070
G gas -19064.5240 6061.1875 857.1813
G solv -3158.7391 64.4568 9.1156
TOTAL -7522.2032 51.2973 7.2545
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1268.1888 14.2342 2.0130
EEL -11557.0773 71.7127 10.1417
EPB -2485.3559 54.5638 7.7165
ECAVITY 47.5088 0.4610 0.0652
G gas -12825.2661 5345.3320 755.9441
G solv -2437.8471 54.5658 7.7168
TOTAL -5118.8075 38.9610 5.5099
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.4198 1.3322
EEL -4684.4720 36.1449 5.1117
EPB -1684.5802 28.2572 3.9962
ECAVITY 28.1687 0.3939 0.0557
G gas -5213.7811 1395.1865 197.3092
G solv -1656.4114 28.2599 3.9966
TOTAL -2313.4381 24.9082 3.5225
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -66.2966 4.2751 0.6046
EEL -959.1803 34.9190 4.9383
EPB 942.7215 33.8861 4.7922
ECAVITY -7.2022 0.3069 0.0434
DELTA G gas -1025.4769 1237.6138 175.0250
DELTA G solv 935.5194 33.8875 4.7924
DELTA G binding = -89.9575 +/- 8.2480 1.1664
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
I21A MUTANT:
POISSON BOLTZMANN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1855.4226 17.0765 2.4150
EEL -17210.2882 75.8866 10.7320
EPB -3229.1405 64.8100 9.1655
ECAVITY 68.5521 0.7596 0.1074
G gas -19065.7108 6050.3755 855.6523
G solv -3160.5884 64.8144 9.1661
TOTAL -7520.1586 50.6710 7.1660
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1261.9126 14.1817 2.0056
EEL -11566.4419 71.5475 10.1183
EPB -2487.5603 54.6289 7.7257
ECAVITY 47.6466 0.4632 0.0655
G gas -12828.3545 5320.1609 752.3844
G solv -2439.9137 54.6309 7.7260
TOTAL -5118.8820 38.4370 5.4358
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.4198 1.3322
EEL -4684.4720 36.1449 5.1117
EPB -1684.5802 28.2572 3.9962
ECAVITY 28.1687 0.3939 0.0557
G gas -5213.7811 1395.1865 197.3092
G solv -1656.4114 28.2599 3.9966
TOTAL -2313.4381 24.9082 3.5225
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -64.2010 4.0841 0.5776
EEL -959.3742 34.9114 4.9372
EPB 942.9999 34.0350 4.8133
ECAVITY -7.2632 0.3107 0.0439
DELTA G gas -1023.5752 1235.4872 174.7243
DELTA G solv 935.7367 34.0364 4.8135
DELTA G binding = -87.8385 +/- 8.0665 1.1408
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
RESULT OF ALANINE SCANNING: (I21A MUTANT:) DELTA DELTA G binding = 2.1190 +/- 11.5368
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
|
该结果文件与前面的同类文件的不同在于, 在每种方法之后会报告结合的ΔΔG, 表明突变对于复合物的结合自由能ΔG的相对影响. 特定的突变也在文件末尾输出. 在本例中, 我们将残基21从异亮氨酸突变为丙氨酸(即I21A).
3.5 分解单个残基或每对残基对Ras-Raf结合自由能的贡献(适用于amber11)
MMPBSA.py计算结合自由能所需的拓扑文件和mdcrd: ras-raf_top_mdcrd.tgz
我们将对3.1节获得的Ras-Raf体系进行结合自由能分解. AMBER支持两种类型的分解方法: 分解到每对残基和分解到单个残基. 单个残基分解方法通过计算单个残基与体系中其他所有残基的相互作用总和来计算单个残基的能量贡献. 成对分解计算体系中残基对之间的相互作用能. 下面将分别展示这两种类型的能量分解. 需要注意的是, 只有 MMPBSA.py正确识别出输入的残基后, 输出的每个残基的DELTA贡献才是有效的.
a. 单个残基的自由能分解
要进行分解, 必须在MMPBSA.py的输入文件中指定&decomp命名列表. 此外, 必须指定idecomp变量(没有默认值). 未能为此变量赋值将导致程序终止, 并给出错误信息. idecomp看指定4个允许的值, 其中两个用于单个残基的分解, 另外两个用于残基对分解. 值1和2决定了单个残基分解的方式: 值1将把1-4非键相互作用能(1-4 EEL和1-4 VDW)加到内部势能项上; 值2将把1-4 EEL相互作用能加到静电势项上, 将1-4 VDW相互作用能加到范德华势能项上.
下面的MMPBSA.py输入文件mmpbsa_per_res_decomp.in使用PB和GB隐式溶剂模型进行单个残基分解:(注意:PB的非极性溶剂化能目前不可分解)
| mmpbsa_per_res_decomp.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | Per-residue GB and PB decomposition
&general
endframe=50, verbose=1,
/
&gb
igb=5, saltcon=0.100,
/
&pb
istrng=0.100,
/
&decomp
idecomp=1, print_res="5; 30-40; 170-200"
dec_verbose=1,
/
|
文件中dec_verbose变量用于指定能量分解输出文件的详细程度.
运行命令如下:
$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -do FINAL_DECOMP_MMPBSA.dat -sp ras-raf_solvated.prmtop -cp ras-raf.prmtop -rp ras.prmtop -lp raf.prmtop -y *.mdcrd
注意, 上述命令可以用MMPBSA.py.MPI并行计算, 详情可参考3.3节.
最终结果文件FINAL_RESULTS_MMPBSA.dat, FINAL_DECOMP_MMPBSA.dat
| FINAL_RESULTS_MMPBSA.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 | | Run on Thu May 20 14:55:43 EDT 2010
|Input file:
|--------------------------------------------------------------
|Per-residue GB and PB decomposition
|&general
| endframe=50, verbose=1,
|/
|&gb
| igb=5, saltcon=0.100,
|/
|&pb
| istrng=0.100,
|/
|&decomp
| idecomp=1, print_res="5; 30-40; 170-200"
| dec_verbose=1,
|/
|--------------------------------------------------------------
|Complex topology file: ras-raf.prmtop
|Receptor topology file: ras.prmtop
|Ligand topology file: raf.prmtop
|Initial mdcrd(s): prod.mdcrd
|
|Best guess for receptor mask: ":1-166"
|Best guess for ligand mask: ":167-242"
|Calculations performed using 50 frames.
|Poisson Boltzmann calculations performed using internal PBSA solver in sander.
|
|All units are reported in kcal/mole.
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
GENERALIZED BORN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1863.7944 16.9979 2.4039
EEL -17200.7297 75.1734 10.6311
EGB -2918.9628 65.1000 9.2065
ESURF 92.2138 0.9782 0.1383
G gas -19064.5240 77.0712 10.8995
G solv -2826.7490 65.1073 9.2076
TOTAL -21891.2730 52.3724 7.4066
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1268.1888 14.0912 1.9928
EEL -11557.0773 70.9920 10.0398
EGB -2314.8693 56.2410 7.9537
ESURF 64.4513 0.6128 0.0867
G gas -12825.2661 72.3770 10.2356
G solv -2250.4181 56.2443 7.9542
TOTAL -15075.6842 36.8322 5.2089
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.3251 1.3188
EEL -4684.4720 35.7816 5.0603
EGB -1587.3051 26.8494 3.7971
ESURF 38.5992 0.5158 0.0730
G gas -5213.7811 36.9768 5.2293
G solv -1548.7058 26.8544 3.7978
TOTAL -6762.4869 26.1943 3.7044
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -66.2966 4.2321 0.5985
EEL -959.1803 34.5681 4.8887
EGB 983.2116 33.0175 4.6694
ESURF -10.8367 0.3832 0.0542
DELTA G gas -1025.4769 34.8262 4.9252
DELTA G solv 972.3749 33.0197 4.6697
DELTA G binding = -53.1020 +/- 6.8437 0.9678
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
POISSON BOLTZMANN:
Complex:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1863.7944 16.9979 2.4039
EEL -17200.7297 75.1734 10.6311
EPB -3216.4587 65.8638 9.3146
ECAVITY 67.8762 0.7739 0.1094
G gas -19064.5240 77.0712 10.8995
G solv -3148.5825 65.8684 9.3152
TOTAL -22213.1066 51.7402 7.3172
Receptor:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -1268.1888 14.0912 1.9928
EEL -11557.0773 70.9920 10.0398
EPB -2489.5955 55.9343 7.9103
ECAVITY 47.1495 0.4689 0.0663
G gas -12825.2661 72.3770 10.2356
G solv -2442.4460 55.9363 7.9106
TOTAL -15267.7121 38.0243 5.3774
Ligand:
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -529.3090 9.3251 1.3188
EEL -4684.4720 35.7816 5.0603
EPB -1673.2574 27.4055 3.8757
ECAVITY 28.0328 0.4091 0.0579
G gas -5213.7811 36.9768 5.2293
G solv -1645.2246 27.4085 3.8761
TOTAL -6859.0057 24.7882 3.5056
Differences (Complex - Receptor - Ligand):
Energy Component Average Std. Dev. Std. Err. of Mean
-------------------------------------------------------------------------------
VDWAALS -66.2966 4.2321 0.5985
EEL -959.1803 34.5681 4.8887
EPB 946.3942 34.1674 4.8320
ECAVITY -7.3062 0.2973 0.0420
DELTA G gas -1025.4769 34.8262 4.9252
DELTA G solv 939.0881 34.1687 4.8322
DELTA G binding = -86.3888 +/- 8.1817 1.1571
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
WARNINGS:
igb=5 should be used with either mbondi2 or bondi pbradii set. Yours are modified Bondi radii (mbondi)
|
| FINAL_DECOMP_MMPBSA.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 | | Run on Thu May 20 14:55:43 EDT 2010
idecomp = 1: Decomposition per-residue adding 1-4 interactions added to Internal.
Energy Decomposition Analysis (All units kcal/mol): Generalized Born solvent
DELTAS:
Total Energy Decomposition:
Residue | Location | Internal | van der Waals | Electrostatic | Polar Solvation | Non-Polar Solv. | TOTAL
-------------------------------------------------------------------------------------------------------------------------------------------------------
LYS 5 | R LYS 5 | 0.000 +/- 4.870 | -0.156 +/- 1.465 | 69.267 +/- 9.154 | -67.061 +/- 9.601 | -0.009 +/- 0.156 | 2.040 +/- 14.208
ASP 30 | R ASP 30 | 0.000 +/- 5.623 | -0.065 +/- 0.961 | -52.559 +/- 11.072 | 52.622 +/- 9.530 | 0.000 +/- 0.084 | -0.003 +/- 15.684
GLU 31 | R GLU 31 | 0.000 +/- 5.174 | -0.247 +/- 1.099 | -79.946 +/- 10.550 | 80.630 +/- 9.693 | -0.227 +/- 0.125 | 0.210 +/- 15.272
TYR 32 | R TYR 32 | 0.000 +/- 4.615 | -0.290 +/- 1.515 | 0.639 +/- 4.431 | -0.076 +/- 3.229 | -0.012 +/- 0.175 | 0.261 +/- 7.327
ASP 33 | R ASP 33 | 0.000 +/- 4.464 | -0.556 +/- 1.073 | -103.116 +/- 5.820 | 103.788 +/- 5.821 | -0.459 +/- 0.094 | -0.343 +/- 9.426
PRO 34 | R PRO 34 | 0.000 +/- 3.388 | -1.829 +/- 0.869 | -3.383 +/- 2.647 | 3.854 +/- 1.944 | -0.308 +/- 0.130 | -1.666 +/- 4.800
THR 35 | R THR 35 | 0.000 +/- 5.702 | -1.829 +/- 1.049 | 0.376 +/- 4.365 | 0.947 +/- 2.028 | -0.204 +/- 0.070 | -0.709 +/- 7.536
ILE 36 | R ILE 36 | 0.000 +/- 4.650 | -2.987 +/- 1.918 | 0.092 +/- 2.149 | 0.991 +/- 0.697 | -0.377 +/- 0.072 | -2.282 +/- 5.515
GLU 37 | R GLU 37 | 0.000 +/- 5.221 | -1.627 +/- 1.388 | -126.728 +/- 6.441 | 120.528 +/- 4.686 | -0.745 +/- 0.048 | -8.573 +/- 9.624
ASP 38 | R ASP 38 | 0.000 +/- 3.750 | -1.583 +/- 1.560 | -104.899 +/- 6.925 | 99.370 +/- 5.710 | -0.254 +/- 0.037 | -7.367 +/- 9.852
SER 39 | R SER 39 | 0.000 +/- 3.447 | -2.184 +/- 1.086 | -13.696 +/- 3.959 | 8.918 +/- 1.800 | -0.504 +/- 0.035 | -7.466 +/- 5.655
TYR 40 | R TYR 40 | 0.000 +/- 4.687 | -4.403 +/- 1.682 | -3.076 +/- 2.884 | 1.652 +/- 1.092 | -0.366 +/- 0.042 | -6.193 +/- 5.858
ARG 170 | L ARG 4 | 0.000 +/- 4.987 | -0.094 +/- 1.646 | -86.951 +/- 10.352 | 82.074 +/- 6.005 | -0.147 +/- 0.073 | -5.118 +/- 13.069
VAL 171 | L VAL 5 | 0.000 +/- 3.812 | -0.183 +/- 1.390 | 2.128 +/- 2.460 | -2.010 +/- 0.555 | 0.000 +/- 0.008 | -0.065 +/- 4.778
PHE 172 | L PHE 6 | 0.000 +/- 4.289 | -0.217 +/- 0.944 | 0.037 +/- 1.743 | 0.132 +/- 0.939 | 0.000 +/- 0.064 | -0.048 +/- 4.818
LEU 173 | L LEU 7 | 0.000 +/- 4.907 | -0.398 +/- 1.241 | -0.940 +/- 3.050 | 1.683 +/- 1.446 | 0.000 +/- 0.022 | 0.345 +/- 6.084
PRO 174 | L PRO 8 | 0.000 +/- 3.433 | -0.188 +/- 1.422 | 2.303 +/- 3.219 | -2.589 +/- 1.289 | 0.000 +/- 0.051 | -0.474 +/- 5.083
ASN 175 | L ASN 9 | 0.000 +/- 4.796 | -1.671 +/- 1.017 | -1.833 +/- 4.740 | 4.535 +/- 2.409 | -0.354 +/- 0.119 | 0.678 +/- 7.233
LYS 176 | L LYS 10 | 0.000 +/- 4.403 | -1.848 +/- 0.810 | -33.879 +/- 7.269 | 36.798 +/- 6.704 | -0.315 +/- 0.107 | 0.756 +/- 10.856
GLN 177 | L GLN 11 | 0.000 +/- 4.261 | -3.791 +/- 1.560 | -1.910 +/- 3.338 | 4.530 +/- 2.016 | -0.359 +/- 0.050 | -1.530 +/- 5.983
ARG 178 | L ARG 12 | 0.000 +/- 6.180 | -2.462 +/- 1.321 | -77.671 +/- 6.496 | 73.669 +/- 4.608 | -0.386 +/- 0.076 | -6.850 +/- 10.167
THR 179 | L THR 13 | 0.000 +/- 4.716 | -1.277 +/- 1.200 | -10.976 +/- 3.020 | 9.344 +/- 0.977 | -0.158 +/- 0.031 | -3.068 +/- 5.810
VAL 180 | L VAL 14 | 0.000 +/- 4.196 | -3.837 +/- 1.389 | -3.014 +/- 2.541 | 2.972 +/- 0.804 | -0.501 +/- 0.041 | -4.379 +/- 5.161
VAL 181 | L VAL 15 | 0.000 +/- 4.333 | -1.791 +/- 1.119 | -3.565 +/- 2.809 | 3.472 +/- 0.656 | -0.155 +/- 0.055 | -2.039 +/- 5.324
ASN 182 | L ASN 16 | 0.000 +/- 4.282 | -1.978 +/- 0.859 | -3.199 +/- 5.507 | 3.645 +/- 2.886 | -0.369 +/- 0.085 | -1.900 +/- 7.598
VAL 183 | L VAL 17 | 0.000 +/- 4.088 | -0.187 +/- 1.149 | 1.057 +/- 4.557 | -0.672 +/- 2.388 | 0.000 +/- 0.073 | 0.199 +/- 6.671
ARG 184 | L ARG 18 | 0.000 +/- 4.797 | -0.183 +/- 1.450 | -90.812 +/- 7.977 | 87.306 +/- 6.336 | -0.335 +/- 0.109 | -4.023 +/- 11.353
ASN 185 | L ASN 19 | 0.000 +/- 5.744 | -0.018 +/- 0.966 | -0.268 +/- 7.498 | 0.303 +/- 4.029 | 0.000 +/- 0.099 | 0.017 +/- 10.315
GLY 186 | L GLY 20 | 0.000 +/- 2.371 | -0.008 +/- 0.701 | -0.334 +/- 2.324 | 0.379 +/- 1.810 | 0.000 +/- 0.057 | 0.037 +/- 3.846
MET 187 | L MET 21 | 0.000 +/- 3.770 | -0.156 +/- 1.254 | -1.692 +/- 3.999 | 1.697 +/- 2.588 | -0.031 +/- 0.089 | -0.181 +/- 6.204
SER 188 | L SER 22 | 0.000 +/- 5.828 | -0.013 +/- 1.008 | 2.808 +/- 4.910 | -2.793 +/- 1.893 | 0.000 +/- 0.061 | 0.002 +/- 7.917
LEU 189 | L LEU 23 | 0.000 +/- 4.943 | -0.021 +/- 1.312 | 1.683 +/- 2.195 | -1.464 +/- 0.671 | 0.000 +/- 0.013 | 0.197 +/- 5.606
HIP 190 | L HIP 24 | 0.000 +/- 5.252 | -0.024 +/- 1.131 | -43.617 +/- 5.567 | 43.652 +/- 4.925 | 0.000 +/- 0.083 | 0.011 +/- 9.172
ASP 191 | L ASP 25 | 0.000 +/- 3.724 | -0.058 +/- 0.723 | 62.413 +/- 8.165 | -61.719 +/- 8.199 | 0.000 +/- 0.107 | 0.636 +/- 12.178
CYS 192 | L CYS 26 | 0.000 +/- 5.318 | -0.098 +/- 1.398 | 1.937 +/- 3.894 | -1.552 +/- 1.485 | 0.000 +/- 0.042 | 0.287 +/- 6.900
LEU 193 | L LEU 27 | 0.000 +/- 4.324 | -0.108 +/- 1.390 | 0.884 +/- 2.119 | -0.740 +/- 0.648 | 0.000 +/- 0.006 | 0.036 +/- 5.054
... 以下250行省略
|
FINAL_DECOMP_MMPBSA.dat输出文件包含有关每个残基与体系其余部分相互作用的信息, 这些信息分为: 内能(idecomp=1时, 势能项包括键, 角, 二面角和1-4相互作用), 范德华能(idecomp=2时, 包括VDW和1-4 VDW), 静电能(idecomp=2时, 包括EEL和1-4 EEL), 极性溶剂化能和非极性溶剂化能. 这个文件被分成如下几个部分:
复合物, 受体, 配体和DELTA(定义为复合物减去受体减去配体)中每个残基的分解能量会分别输出到各自的部分中. 此外, 每个残基的能量贡献会进一步分解为对骨架, 侧链及总能量贡献. Backbone是骨架原子与体系中所有其他原子之间的相互作用能, Sidechain是侧链原子与体系中所有其他原子之间的相互作用能. Total是残基中每个原子与系统中所有其他原子之间的相互作用能(因此是该残基的Backbone和Sidechain值的总和). 每个残基项会分解成上述的组成部分, 由相互作用的平均值+/-该项的标准偏差组成. DELTA部分额外包含一个名为Location的列, 列出复合物中特定残基的位置(R代表受体, L代表配体). 变量dec_verbose控制分解输出文件的详细程度(详细信息请参阅手册).
b. 残基对能量分解
注意: 根据我们的经验, 用PB隐式溶剂模型进行残基对能量分解分析需要非常长的时间才能完成. 下面的分析, 同时使用了GB和PB对50帧进行分析, 在9个处理器(9个独立的32位单核2.8 GHz Xeon处理器)上花费了61小时. 而GB分析只花了3分钟. 所以如果你选择进行PB残基对能量分解, 可能需要很长的模拟时间.
在本节中, 我们将修改输入文件以执行残基对能量分解. 与上面的单个残基能量分解的输入文件大部分相同, 残基对能量分解输入文件mmpbsa_pairwise_decomp.in如下所示:
| mmpbsa_pairwise_decomp.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 | Pairwise GB and PB decomposition
&general
endframe=50, verbose=1,
/
&gb
igb=5, saltcon=0.100,
/
&pb
istrng=0.100,
/
&decomp
idecomp=1, print_res="5; 30-40; 170-200"
dec_verbose=0,
/
|
用于启动MMPBSA.py的命令与单个残基能量分解的命令相同. 但是, 定义用于残基对能量分解的&decomp命名列表中的print_res时需格外小心. 残基对能量分解需要计算的项数为n2, 其中n为print_res指定的残基数. 默认情况下, print_res对应于复合物中的每个残基, 对Ras-Raf会创建大约65 MB(超过450,000行)的分解输出文件. 而且, 由sander创建的mdout文件也将非常大(可能有几个GB, 具体取决于分析的帧数和残基对数), 并且解析所需的内存/时间需求变得相当巨大(即, 解析输出可能需要几分钟时间). 计算的残基对是print_res中指定的残基与print_res中指定的每个其他残基之间的残基对. 有关print_res变量的语法说明, 请参阅手册.
FINAL_DECOMP_MMPBSA.dat的部分输出结果如下图所示:
| FINAL_DECOMP_MMPBSA.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 | | Run on Sun May 23 05:36:28 EDT 2010
idecomp = 3: Pairwise decomposition adding 1-4 interactions added to Internal.
Pairwise Energy Decomposition Analysis (All units kcal/mol): Generalized Born solvent
DELTAS:
Total Energy Decomposition:
Resid 1 | Resid 2 | Internal | van der Waals | Electrostatic | Polar Solvation | Non-Polar Solv. | TOTAL
-----------------------------------------------------------------------------------------------------------------------------------------------------
LYS 5 | LYS 5 | 0.000 +/- 0.000 | 0.000 +/- 1.075 | 0.000 +/- 3.408 | 1.601 +/- 7.229 | 0.000 +/- 0.051 | 1.601 +/- 8.064
LYS 5 | ASP 30 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.341 | -0.000 +/- 0.339 | 0.000 +/- 0.000 | -0.000 +/- 0.480
LYS 5 | GLU 31 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.350 | 0.000 +/- 0.348 | 0.000 +/- 0.000 | 0.000 +/- 0.494
LYS 5 | TYR 32 | 0.000 +/- 0.000 | 0.000 +/- 0.001 | 0.000 +/- 0.071 | 0.000 +/- 0.070 | 0.000 +/- 0.000 | 0.000 +/- 0.099
LYS 5 | ASP 33 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.434 | 0.000 +/- 0.431 | 0.000 +/- 0.000 | 0.000 +/- 0.612
LYS 5 | PRO 34 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.064 | 0.000 +/- 0.064 | 0.000 +/- 0.000 | 0.000 +/- 0.090
LYS 5 | THR 35 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.189 | 0.008 +/- 0.183 | 0.000 +/- 0.000 | 0.008 +/- 0.262
LYS 5 | ILE 36 | 0.000 +/- 0.000 | 0.000 +/- 0.001 | 0.000 +/- 0.120 | 0.021 +/- 0.130 | 0.000 +/- 0.000 | 0.021 +/- 0.177
... 以下1800行省略
ARG 200 | LEU 197 | 0.000 +/- 0.000 | 0.000 +/- 0.423 | 0.000 +/- 0.779 | -0.226 +/- 0.442 | 0.000 +/- 0.022 | -0.226 +/- 0.991
ARG 200 | LYS 198 | 0.000 +/- 0.000 | 0.000 +/- 0.284 | 0.000 +/- 0.793 | 0.237 +/- 0.572 | 0.000 +/- 0.011 | 0.237 +/- 1.018
ARG 200 | VAL 199 | 0.000 +/- 0.000 | 0.000 +/- 0.291 | 0.000 +/- 0.832 | 1.460 +/- 0.587 | 0.000 +/- 0.019 | 1.460 +/- 1.059
ARG 200 | ARG 200 | 0.000 +/- 0.000 | 0.000 +/- 0.562 | 0.000 +/- 3.888 | 14.394 +/- 2.388 | -0.000 +/- 0.035 | 14.394 +/- 4.598
idecomp = 3: Pairwise decomposition adding 1-4 interactions added to Internal.
Pairwise Energy Decomposition Analysis (All units kcal/mol): Poisson Boltzmann solvent
DELTAS:
Total Energy Decomposition:
Resid 1 | Resid 2 | Internal | van der Waals | Electrostatic | Polar Solvation | Non-Polar Solv. | TOTAL
-----------------------------------------------------------------------------------------------------------------------------------------------------
LYS 5 | LYS 5 | 0.000 +/- 0.000 | 0.000 +/- 1.075 | 0.000 +/- 3.408 | 0.670 +/- 7.128 | 0.000 +/- 0.000 | 0.670 +/- 7.974
LYS 5 | ASP 30 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.341 | 0.007 +/- 0.334 | 0.000 +/- 0.000 | 0.007 +/- 0.477
LYS 5 | GLU 31 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.350 | 0.009 +/- 0.344 | 0.000 +/- 0.000 | 0.009 +/- 0.491
LYS 5 | TYR 32 | 0.000 +/- 0.000 | 0.000 +/- 0.001 | 0.000 +/- 0.071 | 0.002 +/- 0.069 | 0.000 +/- 0.000 | 0.002 +/- 0.098
LYS 5 | ASP 33 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.434 | 0.007 +/- 0.423 | 0.000 +/- 0.000 | 0.007 +/- 0.606
LYS 5 | PRO 34 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.064 | -0.000 +/- 0.063 | 0.000 +/- 0.000 | -0.000 +/- 0.090
LYS 5 | THR 35 | 0.000 +/- 0.000 | 0.000 +/- 0.000 | 0.000 +/- 0.189 | 0.024 +/- 0.175 | 0.000 +/- 0.000 | 0.024 +/- 0.257
LYS 5 | ILE 36 | 0.000 +/- 0.000 | 0.000 +/- 0.001 | 0.000 +/- 0.120 | 0.009 +/- 0.102 | 0.000 +/- 0.000 | 0.009 +/- 0.158
LYS 5 | GLU 37 | 0.000 +/- 0.000 | 0.000 +/- 0.008 | 0.000 +/- 1.649 | -0.223 +/- 1.624 | 0.000 +/- 0.000 | -0.223 +/- 2.315
LYS 5 | ASP 38 | 0.000 +/- 0.000 | 0.000 +/- 0.009 | 0.000 +/- 1.802 | -0.191 +/- 1.383 | 0.000 +/- 0.000 | -0.191 +/- 2.272
... 以下1800行省略
|
值得注意的是, FINAL_RESULTS_MMPBSA.dat输出的结果与单个残基能量分解的结果完全相同, 因为能量分解的方式不会影响总值. 因此, 这里忽略了文件以避免重重.
3.6 利用简正模式分析(Nmode)计算雌激素受体和雷洛昔芬复合物的熵
0. 简正模式计算的参数设置
请注意, 截至2010年5月, 简正计算是通过nab编译的nab程序mmpbsa_py_nabnmode完成的, 正常的安装过程可参考这里. 该程序必须存在于PATH或$AMBERHOME/exe中才能运行计算. mmpbsa_py_nabnmode和nmode之间的主要区别在于nab程序可以计算广义波恩(GB)溶剂中的简正模式, 因此另外又增加了两个输入变量(参见手册中的nmode_igb和nmode_istrng). 这个实现还应该改进了早期版本脚本中出现的最小化收敛问题.
1. 构建起始拓扑和坐标文件, 模拟获得平衡体系
方法见3.3节.
2. 简正模式分析计算结合熵(normal mode)
我们将计算复合物, 受体和配体的简正模式, 并对结果进行平均以估计结合熵. 请注意, 复合物体系的熵贡献可以通过取消&general命名列表中最后一行的注释进, 利用AmberTools中的ptraj模块进行准简谐熵计算.
我们将利用MMPBSA.py的输入文件mmpbsa.in进行简正模式计算:
| mmpbsa.in | |
|---|---|
1 2 3 4 5 6 7 8 | Input file for running entropy calculations using NMode
&general
endframe=50, keep_files=2,
/
&nmode
nmstartframe=1, nmendframe=50,
nminterval=5, nmode_igb=1, nmode_istrng=0.1,
/
|
文件中的endframe变量设置当用ptraj构建无溶剂体系的mdcrd文件时, 读入轨迹时在哪一帧停止. keep_files变量允许用户指定计算结束后删除哪些文件. &nmode命名列表用于定义用于简正模式计算的变量. 对要分析的无溶剂体系, nmstartframe变量定义简正模式分析开始时对应的帧. nmendframe和nminterval分别定义了简正模式分析的结束帧和帧间隔. 值得注意的是, nmstartframe/endframe/interval这些变量对应的”轨迹”是由&general命名列表中定义的startframe, endframe和interval变量提取出来的快照. 因此, 只有这些帧的一部分才能用于简正模式计算(最多是_MMPBSA_complex.mdcrd中的每帧).
运行命令如下:
$AMBERHOME/bin/MMPBSA.py -O -i mmpbsa.in -o FINAL_RESULTS_MMPBSA.dat -sp 1err.solvated.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y *.mdcrd > progress.log
或者使用下面的命令将作业脚本提交到排队系统, 如PBS系统:
qsub nmode.job
作业脚本nmode.job看上去和bash类似, 内容如下:
| nmode.job | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 | #!/bin/sh
#PBS -N nmode
#PBS -o nmode.out
#PBS -e nmode.err
#PBS -m abe
#PBS -M email@domain.edu
#PBS -q brute
#PBS -l nodes=1:surg:ppn=3
#PBS -l pmem=1450mb
cd $PBS_O_WORKDIR
mpirun -np 3 MMPBSA.py.MPI -O -i mmpbsa_nm.in -o FINAL_RESULTS_MMPBSA.dat -sp 1err.solvated.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y 1err_prod.mdcrd > progress.log
|
计算进度会输出到文件progress_nm.log. 计算过程中的所有错误也会输出到这个文件. 最后, 计算完成后会显示每个步骤所用的时间. 如果串行计算10帧大约需要40个小时. MMPBSA.py.MPI可以用来并行(运行细节请参考3.3节). 计算时会根据线程数量平均分配帧数. 体系的大小对计算时间和所需内存(RAM)有显著影响. 段错误(segfaults)通常是由于系统中RAM不足造成的.
| progress_nm.log | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | ptraj found! Using /share/local/lib/amber10/i686/bin/ptraj
sander found! Using /share/local/lib/amber10/i686/bin/sander (serial only!)
nmode found! Using /share/local/lib/amber10/i686/bin/nmode
Preparing trajectories with ptraj...
50 frames were read in and processed by ptraj for use in calculation.
Starting sander calls
Starting nmode calculations...
Timing:
Processing Trajectories With Ptraj: 0.240 min.
Total Harmonic nmode Calculation Time: 2363.906 min.
Output File Writing Time: 0.018 min.
Total Time Taken: 2364.165 min.
MMPBSA Finished. Thank you for using. Please send any bugs/suggestions/comments to mmpbsa.amber@gmail.com
|
输出文件Nmode_output.tgz
上述命令利用ptraj生成了三个非溶剂化mdcrd文件(复合物, 受体和配体), 其中包含计算熵变时分析过的坐标. 简正模式计算时利用每个快照的PDB文件, 因此从复合物, 受体和配体的无溶剂mdcrd文件生成了10个PDB文件. 如果entropy=1, 还会生成一个平均结构的PDB文件, 其中的结构(通过RMS)对齐到所有快照. 如果需要, 可以使用ptraj对这个结构进行准简谐熵计算.
最终结果文件FINAL_RESULTS_MMPBSA.dat:
| FINAL_RESULTS_MMPBSA.dat | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 | | Run on Thu Feb 11 12:44:26 EST 2010
|Input file:
|--------------------------------------------------------------
|Input file for running entropy calculations using NMode
|&general
| endframe=50, keep_files=2,
|/
|&nmode
| nmstartframe=1, nmendframe=50,
| nminterval=5,
|/
|--------------------------------------------------------------
|Solvated complex topology file: 1err.solvated.prmtop
|Complex topology file: complex.prmtop
|Receptor topology file: receptor.prmtop
|Ligand topology file: ligand.prmtop
|Initial mdcrd(s): 1err_prod.mdcrd
|
|Best guess for receptor mask: ":1-240"
|Best guess for ligand mask: ":241"
|Ligand residue name is "RAL"
|
|Calculations performed using 50 frames.
|Poisson Boltzmann calculations performed using internal PBSA solver in sander.
|
|All units are reported in kcal/mole.
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
ENTROPY RESULTS (HARMONIC APPROXIMATION) CALCULATED WITH NMODE:
Complex:
Entropy Term Average Std. Dev.
-----------------------------------------------------------
Translational: 16.9389 0.0000
Rotational: 17.3953 0.0038
Vibrational: 2784.7967 2.3982
Total: 2819.1307 2.3964
Receptor:
Entropy Term Average Std. Dev.
-----------------------------------------------------------
Translational: 16.9233 0.0000
Rotational: 17.3911 0.0045
Vibrational: 2755.5693 3.0352
Total: 2789.8840 3.0342
Ligand:
Entropy Term Average Std. Dev.
-----------------------------------------------------------
Translational: 13.2972 0.0000
Rotational: 11.4991 0.0496
Vibrational: 33.7549 0.0442
Total: 58.5511 0.0058
DELTA S total= -30.0192 +/- 3.4064
NOTE: All entropy results have units kcal/mol. (Temperature has already been multiplied in as 300. K)
-------------------------------------------------------------------------------
-------------------------------------------------------------------------------
|
该输出文件的开始部分包含有关计算的各种细节. 输出文件的其余部分包括平动, 转动, 振动和总熵贡献的平均值, 标准偏差和均值的标准误差. 在这些部分之后, ΔS以均值以及标准差和标准误差的形式给出.
通常我们会期望生物复合物的ΔS值为负. 这意味着蛋白质和配体结合形成复合物时可用微观状态数减少. 可用微观状态数的减少主要来自配体被限制在结合位点附近, 并且当配体与蛋白结合之后, 配体的运动受到了限制.
我们得到的ΔS值等于-30.02 kcal/mol, 这意味着在纯水中该蛋白-配体复合体的生成是一个熵不利的过程. 但是, 该结果不等于真实的结合自由能, 因为我们没有估计结合自由能中的(有利的) 焓贡献.
