- 先决条件
- 内容
- Barnett GROMACS教程1: 水
- Barnett GROMACS教程2: 水中的单个甲烷
- Barnett GROMACS教程3: 水中的多个甲烷
- Barnett GROMACS教程4: 甲烷的溶剂化自由能
- Barnett GROMACS教程5: 使用伞形采样计算甲烷-甲烷的平均力势PMF
- Barnett GROMACS教程6: 使用测试粒子插入计算甲烷的超额化学势
- 2018-11-24 21:02:36
注意: 我将在2018年底撤下这些教程. 我没法花更多时间来维护它们并保持更新. 你仍然可以通过Internet Archive的Wayback Machine获取这些教程.
这些教程是我在博士期间写的一些GROMACS入门教程. 我发现很多GROMACS教程侧重于生物分子, 如蛋白质, 很少有人关注简单的系统. 我希望这些教程能填补这个空白, 让你更好地理解如何设置和运行简单的分子模拟, 以及如何利用先进的采样技术.
没有必要按顺序完成教程, 但是在继续其他教程之前, 前两个教程是必需的, 因为所有后续教程中都使用了教程2中甲烷的结构文件(methane.pdb)和拓扑文件(topol.top). 本教程是为GROMACS 5.1及以上版本设计的. 如果你使用的是旧版本, 则某些命令或参数可能已更改. 请特别注意, 自GROMACS 5.0版本发布以来, 伞形采样的牵引代码已发生变化.
先决条件
我假设你对命令行(例如, bash)操作有一定的知识. 具体来说, 你应该知道如何创建目录, 改变目录, 编辑文本文件以及将文件下载到系统中. 当你看到$或>时, 这是命令行提示符, 指示你应该在它后面键入文本. 如果对于命令行你是新手, 请考虑搜索教程并学习一下.
我还假设你已经在可用的机器上安装了GROMACS. 源代码和安装说明可以在GROMACS手册上找到.
在整个教程中, 我们将使用OPLS甲烷和TIP4PEW水.
内容
- 水 - 设置模拟的基础知识. 确定TIP4PEW水的密度.
- 水中的单个甲烷 - 如何为分子创建拓扑文件并进行溶剂化. 获得径向分布函数.
- 水中的几个甲烷 - 如何将多个溶质分子加入系统中. 获得甲烷-甲烷的平均力势.
- 甲烷的溶剂化自由能 - 耦合分子时如何进行自由能模拟. 使用MBAR获得结果.
- 伞形采样 - 使用牵引代码从伞形采样中获取甲烷-甲烷的平均力势.
- 测试粒子插入 - 使用测试粒子插入获得甲烷的超额化学势.
Barnett GROMACS教程1: 水
在本入门教程中, 我将向您展示如何创建一个水盒子, 并在恒定的温度和压力下对其进行简单模拟. 模拟完成后我们就可以确定出水的密度.
设置
每一GROMACS模拟都需要三个基本文件: 结构文件(.gro/.pdb), 拓扑文件(.top)和参数文件(.mdp). 结构文件包含系统中每个原子的直角坐标. 拓扑文件包含每个原子如何与其他原子相互作用的信息, 这里所说的相互作用既包括非键相互作用也包括成键相互作用. 这些相互作用的信息由力场提供. 非键相互作用包括范德华相互作用和库仑相互作用. 成键相互作用包括键长, 键角和二面角. 参数文件包含模拟的运行时间, 时间步长, 温度和压力耦合等信息. 下面我们将获取/创建这些文件.
我建议创建一个目录来存储本教程的文件.
拓扑文件
我们将从拓扑文件开始. 通常, 拓扑文件使用#include语句来引用要使用的力场. 其中包括[ atomtypes ], [ bondtypes ], [ angletypes ]和[ dihedraltypes ]指令. 然后, 在拓扑文件中, 我们通常还会指定不同的[ moleculetype ]指令, 其中又包含了[ atoms ], [ bonds ]和[ dihedrals ]指令, 它们都来自于所引用的力场. 现在不要太担心这些. 因为水模型已经为我们准备好了所有需要的这些信息. 更多信息情况请参考手册第5章.
创建一个名为topol.top的文件, 其文本如下:
| topol.top | |
|---|---|
1 2 3 4 5 6 | #include "oplsaa.ff/forcefield.itp"
#include "oplsaa.ff/tip4pew.itp"
[ System ]
TIP4PEW
[ Molecules ] |
如你所见, 我们已经引用了OPLS-AA力场. 此外, 我们还引用了TIP4PEW水模型. 紧接着是一个[ System ]指令, 其中只包含系统的名称, 可以是任意字符. 最后, 我们在[ Molecules ]下列出每种分子的类型及其数目. 现在我们还没有这些信息(不过我们马上就会知道).
结构文件
GROMACS在拓扑目录中提供了TIP4PEW水模型的结构文件. 拓扑目录的标准位置通常是/usr/share/gromacs/top, 但也可能被你放到了不同的目录. 如果你可以正确地执行source GMXRC, 那么拓扑目录将位于$GMXDATA/top. 在该目录中, 您会看到几个.gro文件, 其中之一是tip4p.gro. 您还会看到上面的拓扑文件中引用的文件夹oplsaa.ff. TIP4PEW没有专用的结构文件. 因为TIP4P和TIP4PEW这两个四位点水模型的结构基本相同. 它们的不同之处在于力场参数.
要使用结构文件创建水盒子, 请执行以下操作:
$ gmx solvate -cs tip4p -o conf.gro -box 2.3 2.3 2.3 -p topol.top如果你打开topol.top, 会看到文件最后添加了一行, 内容是SOL和一个数字. SOL是在oplsaa.ff/tip4pew.itp中定义的moleculetype的名称. 当我们运行上面的gmx solvate命令时, GROMACS会在边长为2.3 nm的盒子中添加足够的水分子.
参数文件
现在我们需要一组参数文件, 这样GROMACS才能知道如何处理我们的起始结构. 模拟几乎总是包含三个主要阶段: 能量最小化, 预平衡和成品模拟. 其中的能量最小化和预平衡又可以分解为多个步骤. 其中的每一步都需要单独的参数文件. 在本教程中, 我们将进行两次能量最小化, 两次预平衡和一次成品模拟.
我们要使用的参数文件可以从这里下载.
这五个文件有一些相同的内容. 对每个选项, 我只给出了一个简短的注释. 有关每个选项的更多信息, 请参阅GROMACS网站
| 参数 | 值 | 解释说明 |
|---|---|---|
| cutoff-scheme | Verlet | 创建邻近列表的方法. 当前GROMACS版本的默认设置, 在这里明确指定以避免GROMACS给出注意信息 |
| coulombtype | PME | 对长程(k-space)静电使用Particle-Mesh Ewald方法 |
| rcoulomb | 1.0 | PME计算中实空间(k空间)的截断距离, 单位nm |
| vdwtype | Cut-off | 范德华力在rvdw处截断 |
| rvdw | 1.0 | VDW的截断距离, 单位nm |
| DispCorr | EnerPress | 对VDW能量和压力都进行长程校正 |
在设定截断距离时, 要考虑到力场是如何参数化的. 换句话说, 最好查看下相关力场的论文, 看它是如何创建的. 我们在这里选择1.0 nm作为截断距离, 对于OPLS来说已经足够了, 但您也可以为自己的系统选择其他值.
此外, 在每次模拟中, 我们还将输出能量文件, 日志文件和压缩轨迹文件. 它们(在模拟步骤中)的输出频率分别使用nstenergy, nstlog和nstxout-compressed进行设置. 运行成品模拟时我们将输出更多信息.
除了第二次能量最小化之外, 对于每次模拟, 我们还将设置constraint-algorithm = lincs和constraints = h-bonds, 来使用LINCS算法约束所有涉及氢原子的键. 这样我们就可以使用更大的时间步长.
对于第一次能量最小化, 我们设置integrator = steep来使用最陡下降算法对体系能量进行最小化, 最大步数为1000步(nsteps = 1000). 如果在此之前能量收敛, 最小化将停止. 此外, 我们还使用了define = -DFLEXIBLE. 这样GROMACS会使用柔性水模型, 因为默认情况下所有的水模型都是使用SETTLE算法的刚性模型. 在我们引用的水模型的拓扑文件中, 有一个if语句, 用于查找要定义的FLEXIBLE变量. 第一次能量最小化的目的是调整分子的初始位置, 这样使用SETTLE约束算法时就不容易遇到错误了.
在第二次能量最小化的参数文件中, 我们只需要删除define = -DFLEXIBLE并将最大步数增加到50,000即可.
最后三次模拟-两次预平衡模拟和成品模拟-都设置integrator = md来使用蛙跳式积分方法. 此外, 设置dt=0.002来使用2 fs时间步长.
对于第一次预平衡, 有几点需要注意. 我们增加了几个参数, 如下所示:
| 参数 | 值 | 解释说明 |
|---|---|---|
| gen-vel | yes | 根据Maxwell-Boltzmann分布为每个原子产生速度, 这样体系的初始温度会接近于我们将要耦合的温度. 只需要为第一次预平衡步骤产生速度 |
| gen-temp | 298.15 | gen-vel使用的温度, 单位K. 除非你需要做一些奇怪/有趣的事情, 否则设定值应该与ref-t相同 |
| tcoupl | Nose-Hoover | 温度耦合算法. Nose-Hoover算法可以正确地生成正则系综 |
| tc-grps | System | 要耦合的组. 你可以分别耦合不同的原子组, 但我们这里将整个系统耦合在一起 |
| tau-t | 2.0 | 耦合的时间常数. 详细信息请参阅手册 |
| ref-t | 298.15 | 以K为单位的温度 |
| nhchainlength | 1 | 蛙跳式算法只支持1, 但默认情况下为10. 这样设置是为了避免GROMACS显示警告信息 |
第一次预平衡的目的在于使得体系在加压耦合之前达到正确的温度(298.15 K). 因为如果同时添加温度和压力耦合可能会导致系统不稳定并发生崩溃. 我们不想在模拟一开始出现这种情况. 此外, 我们还设置了nSteps=50000, 使用2 fs的时间步长, 这意味着模拟将运行100 ps. 这对于本教程中的水盒子足够了, 但对于更大/更复杂系统, 你可能需要进行更长时间的预平衡.
第二次预平衡增加了压力耦合. 请注意, 我们并没有再次为原子产生速度, 因为那将使我们前面所做的预平衡失去意义. 我们还为约束设置了continuation = yes, 因为我们从第一次平衡开始继续进行模拟. 这次模拟将运行1 ns. 同样, 对于其他系统, 可能需要更长的模拟时间.
| 参数 | 值 | 解释说明 |
|---|---|---|
| pcoupl | Parrinello-Rahman | 压力耦合算法. 当与Nose-Hoover一起使用时, Parrinello-Rahman能正确地产生等压等温系综 |
| tau-p | 2.0 | 耦合的时间常数. 详细信息请参阅手册 |
| ref-p | 1.0 | 耦合压力, 单位bar |
| compressibility | 4.46e-5 | 系统压缩系数, 单位 bar-1 |
对于成品模拟, 除了输出更多数据和运行10 ns之外, 一切都与最后一次平衡完全相同.
模拟
现在我们已经有了运行每次模拟所需的所有文件. 通常每次模拟运行时都需要先使用gmx grompp将已有的三个文件(.gro, .top和.mdp)预处理为一个.tpr文件(有时也会被称为拓扑文件, 虽然这容易引起混淆).
能量最小化
首先, 执行以下操作来运行两次能量最小化步骤:
$ gmx grompp -f mdp/min.mdp -o min -pp min -po min
$ gmx mdrun -deffnm min
$ gmx grompp -f mdp/min2.mdp -o min2 -pp min2 -po min2 -c min -t min
$ gmx mdrun -deffnm min2对于每次操作, 我们都使用-f选项读取.mdp文件. 默认情况下, 如果未指定-c和-p选项, GROMACS将使用conf.gro和topol.top作为结构和拓扑文件. 另外, 我们使用-pp和-po输出处理过的拓扑文件和mdp文件. 这并非必要, 但值得一看, 尤其是处理过的mdp文件, 因为里面带有注释.
在接下来的每个步骤中, 我们都使用-c和-t选项读取前一步骤的最后一个结构文件或检查点文件. 默认情况下, GROMACS会每15分钟输出一次检查点文件, 最后一步也会输出检查点文件. 如果不存在检查点文件, GROMACS会使用由-c定义的结构文件, 因此最好同时指定这两个文件. 每次运行gmx mdrun时, 我们指示GROMACS为每个输入文件和输出文件使用默认名称, 因为输出文件有多个.
注意进行第二次能量最小化时我们使用了-maxwarn 1选项. 使用这个选项时要小心, 除非你知道自己在做什么. 对我们的情况, GROMACS给出的是关于L-BFGS效率的警告信息, 因此我们可以安全地忽略它.
为了解能量最小化的过程, 让我们使用GROMACS命令gmx energy提取这两次模拟的势能. 执行以下操作并输入与Potential对应的数字, 然后再次输入:
$ gmx energy -f min.edr -o min-energy.xvg现在对第二次能量最小化进行相同的操作:
$ gmx energy -f min2.edr -o min2-energy.xvg生成的.xvg文件的头部包含了供Grace绘图程序使用的信息. 我偏爱使用gnuplot绘图, 因此文件头部的一些行将导致错误. 简单地将.xvg文件中的每个@字符替换为#后, 我就可以使用gnuplot绘图了. 首先启动gnuplot:
$ gnuplot然后在gnuplot终端上执行:
> plot 'min-energy.xvg' w l要绘制第二次能量最小化过程中的能量变化, 请执行以下操作:
> plot 'min2-energy.xvg' w l第一次的结果类似如下:
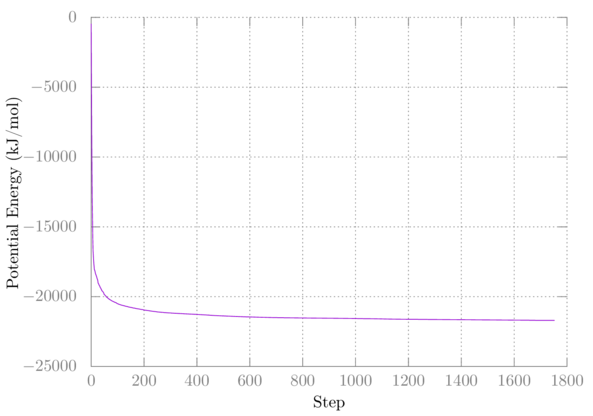
我的第二次能量最小化过程中结果没有任何变化, 所以我就不再绘图了.
预平衡1(NVT)
现在我们已经调整好了初始结构, 让我们添加温度耦合进行第一次预平衡:
$ gmx grompp -f mdp/eql.mdp -o eql -pp eql -po eql -c min2 -t min2
$ gmx mdrun -deffnm eql让我们看看整个模拟过程中温度的变化情况:
$ gmx energy -f eql.edr -o eql-temp.xvg在提示符下选择与Temperature相对应的数字, 回车, 然后再次回车完成选择. 使用gnuplot绘图, 方法如前所示. 您应该看到类似如下的内容:
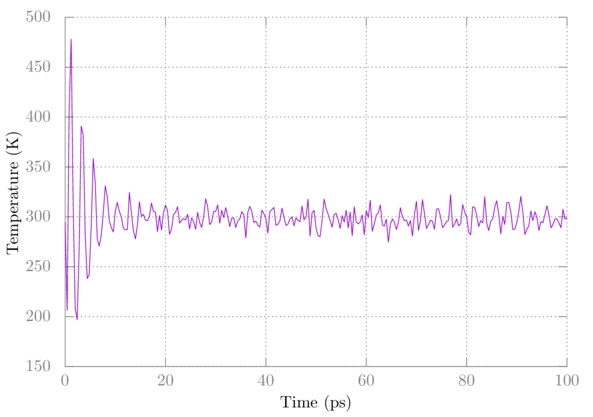
请注意, 温度最初波动很大但最终会稳定下来.
预平衡2(NPT)
对于最后的预平衡, 如前所述, 我们增加了压力耦合:
$ gmx grompp -f mdp/eql2.mdp -o eql2 -pp eql2 -po eql2 -c eql -t eql
$ gmx mdrun -deffnm eql2你可以使用gmx energy检查温度和压力, 方法如前所述. 下面是压力的变化图:
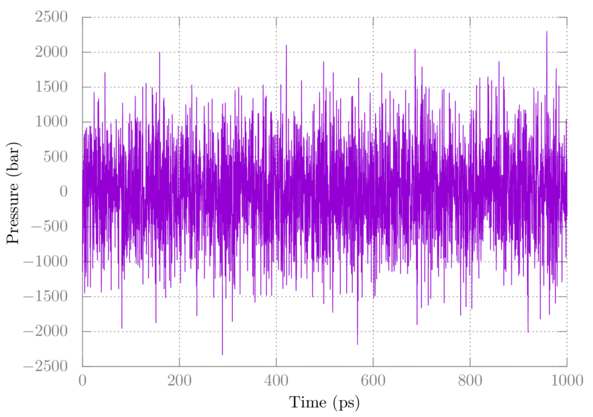
请注意, 压力波动很大, 这是正常的. 在本教程中, 完全平衡后压力的平均值应接近1巴.
成品模拟
现在进行成品模拟:
$ gmx grompp -f mdp/prd.mdp -o prd -pp prd -po prd -c eql2 -t eql2
$ gmx mdrun -deffnm prd分析
使用gmx energy处理prd.edr, 得到平均温度, 压力和密度. 它们的值符合预期么?
以下是我得到的输出:
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Temperature 298.145 0.019 8.65629 0.0338992 (K)
Pressure 3.25876 0.97 688.616 -2.75083 (bar)
Density 995.381 0.15 12.92 0.0705576 (kg/m^3)
如果你看一下TIP4PEW论文中的图4, 就会发现我们已经得到了正确的密度. 另外请注意, Wolfram Alpha中标准条件下水的密度为997 kg /立方米.
您也可以使用像vmd这样的程序来可视化模拟过程. 要使用vmd打开成品模拟的轨迹, 请执行以下操作:
$ vmd prd.gro prd.xtc下面是一个快照:
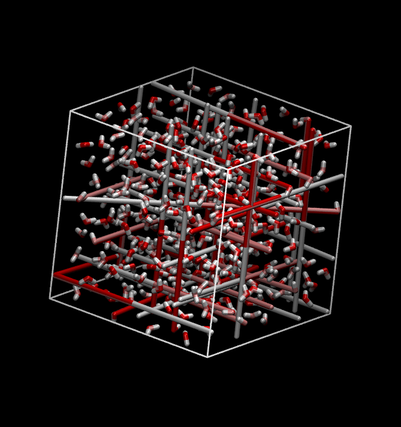
注意, 由于周期性边界条件, 可能会存在贯穿整个盒子的键, 这看起来有些奇怪, 但并不意味着模拟有问题. 你可以使用gmx trjconv完整化分子:
$ gmx trjconv -f prd.xtc -s prd.tpr -pbc mol -o prd-mol.xtc处理后的文件看起来更好些:

总结
在本教程中, 我们使用gmx solvate生成了TIP4PEW水盒子. 并分五个不同的阶段对其进行了: 能量最小化1, 能量最小化2, 预平衡1, 预平衡2和成品模拟. 每次模拟都使用了单独的.mdp文件, 我们对这些文件进行了解释. 对每次模拟, 我们使用gmx energy来提取模拟过程中的有用信息. 成品模拟运行完成后, 我们得到了TIP4PEW水的密度.
Barnett GROMACS教程2: 水中的单个甲烷
在本教程中, 我将向您展示如何在一个TIP4PEW水盒子中创建一个包含单个OPLS力场甲烷的系统.
设置
和以前一样, 我们需要一个结构文件, 一个拓扑文件和几个参数文件. 我们将使用GROMACS工具gmx pdb2gmx从pdb文件生成拓扑.
为pdb2gmx构建残基
对于甲烷分子, 我们将使用OPLS力场. 它位于顶层力场目录(可能是/usr/share/gromacs/top或类似的目录).
如果您不确定GROMACS的安装位置, 可以使用如下命令查看:
echo $GMXPREFIX如果您正确source了GROMACS配置文件, 上述命令可以为您提供GROMACS的安装目录. 在该目录中找到share/gromacs/top并进入(例如, 如果GMXPREFIX是/usr则转到/usr/share/gromacs/top). 也可以直接转到$GMXDATA/top.
让我们看一下力场目录的内容:
cd oplsaa.ff
ls您会看到一些文件, 但我们现在只对其中的几个感兴趣. 注意forcefield.itp. 这是模拟中使用的主要文件. 在里面你会看到[ defaults ]以及对两个其他文件的引用 - 一个用于成键相互作用, 一个用于非键相互作用. 我们还对atomtypes.atp以及aminoacids.rtp感兴趣, 前者对难理解的opls_####项进行了说明, 而后者则给出了gmx pdb2gmx命令能够识别的残基的列表.
用文本编辑器打开atomtypes.atp. 例如可使用vim打开它:
vim atomtypes.atp转到包含opls_138的行. 注意到注释为alkane CH4, 所以我们的甲烷会用到这个原子类型. 但是, 请注意第二列中的质量 - 它只是CH4基团中碳原子的质量, 因此我们还需要氢原子的原子类型. 我们使用的是”全原子”模型 - 每个原子都要指定原子类型. 相应氢原子的类型是opls_140. 你可能需要查看下OPLS力场论文的支撑材料. 论文中的参数应该与我们稍后看到的参数匹配. 现在记下这两种原子类型并关闭文件.
让我们来看看这两种原子类型的ffnonbonded.itp:
grep opls_138 ffnonbonded.itp
grep opls_140 ffnonbonded.itp在这里, 我们可以看到原子类型的名称, 键类型, 质量, 电荷, ptype, sigma和epsilon. 记下每个原子类型的电荷 - 我们需要它们来构建新的残基类型. 另外注意, ffbonded.itp中对键类型, 角度类型和二面角类型统一使用键类型进行表征.
在继续之前, 您可能希望将顶级力场目录复制到别的位置, 比如您的主目录, 因为我们将对其进行修改并添加一些文件. 要将其复制到主目录, 请执行以下操作:
cp -r $GMXDATA/top $HOME/GMXLIB你可能需要root权限才能执行. 现在将$GMXLIB环境变量更改为:
export GMXLIB=$HOME/GMXLIB将上述内容添加到.bash_profile中以便永久有效. 现在执行:
cd $GMXLIB您现在位于目录的副本中, 所有模拟都将使用该目录而不是GROMACS默认目录中提供的目录.
现在进入oplsaa.ff并打开aminoacids.rtp. 您会注意到文件中已有一些残基. 我们将为甲烷添加一个名为methane.rtp的新文件, 其中包含我们称之为CH4的残基. 关闭aminoacids.rtp. 我们需要告诉gmx pdb2gmx残基中的原子及其之间的成键. 我们也可以指定键角, 但无须这么做, 因为gmx pdb2gmx会自动生成键角列表. 使用以下内容创建文件, 并将其另存methane.rtp, 放在oplsaa.ff目录中:
| methane.rtp | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 | [ bondedtypes ]
; bonds angles dihedrals impropers all_dihedrals nrexcl HH14 RemoveDih
1 1 3 1 1 3 1 0
; Methane
[ CH4 ]
[ atoms ]
C opls_138 -0.240 1
H1 opls_140 0.060 1
H2 opls_140 0.060 1
H3 opls_140 0.060 1
H4 opls_140 0.060 1
[ bonds ]
C H1
C H2
C H3
C H4 |
关于上述文件的一些注释: [ bondedtypes ]来自aminoacids.rtp并且是必需的. [ atoms ]下的名称可以随意设置, 只要它们与我们稍后将要创建的PDB文件匹配即可. 请注意, 在第一列中我们给出了原子名称, 原子类型, 电荷, 然后是电荷组. 在[ bonds ]下, 我们只指定了每个原子如何连接到其他原子. 也就是, C与每个氢连接. 我们可以选择性地添加[ angles ], 但如前所述, GROMACS会为我们自动生成. 现在关闭文件. 有关这方面的更多信息, 请参见GROMACS手册第5.6节.
创建pdb文件并运行gmx pdb2gmx
现在我们准备创建pdb文件了. 有一些程序可以创建分子结构文件. 例如Avogadro. 另一种方法是使用AmberTools程序包中的xleap. 在Avogadro中, 只要在窗口中随便点击一下, 就可以得到甲烷. 将此文件另存为methane.pdb. 你的文件应该是这样的. 将它保存在您的主目录中, 但不能保存在$GMXLIB中的任何位置.
将methane.pdb中的LIG更改为CH4. 同时将第一个H更改为H1, 将第二个更改为H2, 依此类推. PDB文件是固定格式, 因此请保证每列的开头处于同一位置. 不需要CONNECT和MASTER记录, 因此可以删除它们. 接下来将UNNAMED改为METHANE. 修改后的文件应该如下所示
| methane.pdb | |
|---|---|
1 2 3 4 5 6 7 8 | COMPND METHANE
AUTHOR GENERATED BY OPEN BABEL 2.3.2
HETATM 1 C CH4 1 -0.370 0.900 0.000 1.00 0.00 C
HETATM 2 H1 CH4 1 0.700 0.900 0.000 1.00 0.00 H
HETATM 3 H2 CH4 1 -0.727 0.122 0.643 1.00 0.00 H
HETATM 4 H3 CH4 1 -0.727 0.731 -0.995 1.00 0.00 H
HETATM 5 H4 CH4 1 -0.727 1.845 0.351 1.00 0.00 H
END |
将文件另存为methane.pdb.
现在我们可以使用gmx pdb2gmx来创建GROMACS结构文件和拓扑文件了:
$ gmx pdb2gmx -f methane.pdb系统将提示您选择力场. 选择OPLS. 如果提示中的OPLS力场有两个不同的目录, 请选择您所复制的目录中的OPLS. 对于水模型选择TIP4PEW. 如果GROMACS给出找不到CH4残基的错误提示, 那么您可能使用了错误的力场.
上述命令将创建三个文件: conf.gro, posre.itp和topol.top. conf.gro是包含单个甲烷的结构文件, topol.top是系统的拓扑文件, posre.itp是溶质(甲烷)的位置限制文件. 最后一个文件并非必需, 我们不会使用它. 在topol.top文件中, 注意到有[ angers ], 正如前面所说, 这是GROMACS自动为我们创建的. 您还需要在topol.top中对甲烷分子进行重命名. 看一看并浏览下每个文件. GROMACS手册第5章可以帮助您更多地了解拓扑文件.
注意: 在其他教程中会再次使用topol.top和methane.pdb文件.
对于那些使用gmx pdb2gmx为蛋白质大分子生成拓扑的人来说, 事情会变得更加复杂. 本教程只是一个简单的例子, 实际上我们可以在其他地方找到需要的拓扑文件.
溶剂化系统
到目前为止, 我们的结构文件和拓扑文件中只有甲烷. 我们需要使用gmx solvate来添加水分子:
$ gmx solvate -cp conf.gro -o conf.gro -cs tip4p -p topol.top -box 2.3 2.3 2.3参数文件
我们将使用与上一教程相同的参数文件.
模拟
我们将使用与上一教程相同的模拟流程. 假设您的mdp参数文件位于名为mdp的目录中:
$ gmx grompp -f mdp/min.mdp -o min -pp min -po min
$ gmx mdrun -deffnm min
$ gmx grompp -f mdp/min2.mdp -o min2 -pp min2 -po min2 -c min -t min
$ gmx mdrun -deffnm min2
$ gmx grompp -f mdp/eql.mdp -o eql -pp eql -po eql -c min2 -t min2
$ gmx mdrun -deffnm eql
$ gmx grompp -f mdp/eql2.mdp -o eql2 -pp eql2 -po eql2 -c eql -t eql
$ gmx mdrun -deffnm eql2
$ gmx grompp -f mdp/prd.mdp -o prd -pp prd -po prd -c eql2 -t eql2
$ gmx mdrun -deffnm prd提示: 您可以将上述命令放在名为run的bash脚本中一起执行. 将以下内容添加到脚本的头部:
#!/bin/bash
set -e然后执行:
$ chmod +x run要运行脚本, 请执行:
$ ./run您的脚本应该如下所示. 其中的set-e告诉bash在出现错误时停止脚本.
| 2-run.bsh | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 | #!/bin/bash
set -e
gmx grompp -f mdp/min.mdp -o min -pp min -po min
gmx mdrun -deffnm min
gmx grompp -f mdp/min2.mdp -o min2 -pp min2 -po min2 -c min -t min
gmx mdrun -deffnm min2
gmx grompp -f mdp/eql.mdp -o eql -pp eql -po eql -c min2 -t min2
gmx mdrun -deffnm eql
gmx grompp -f mdp/eql2.mdp -o eql2 -pp eql2 -po eql2 -c eql -t eql
gmx mdrun -deffnm eql2
gmx grompp -f mdp/prd.mdp -o prd -pp prd -po prd -c eql2 -t eql2
gmx mdrun -deffnm prd |
分析
让我们计算一下所谓的径向分布函数. 首先, 我们需要创建一个索引文件:
$ gmx make_ndx -f conf.gro
> a C
> a OW
> q然后运行gmx rdf:
$ gmx rdf -f prd.xtc -n index.ndx提示时, 选择C作为参考组. 然后选择OW. 然后键入CTRL-D以结束选择. 通过在gnuplot中执行以下操作, 可以绘制数据的第1列和第3列作为结果:
> plot 'rdf.xvg' u 1:3 w l它应该类似下图:
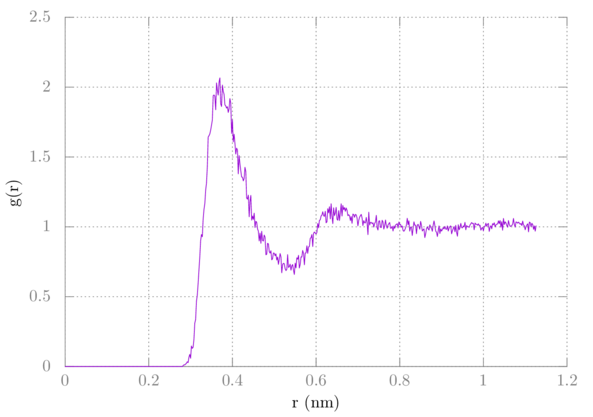
总结
在本教程中, 我们学习了如何创建用于gmx pdb2gmx的残基模板文件(.rtp). 我们创建了OPLS甲烷的结构, 并为其生成了拓扑文件. 然后我们使用gmx solvated为它添加了水溶剂. 在此之后我们进行了模拟, 就像上次一样. 最后, 我们使用gmx rdf计算了C-OW的径向分布函数.
Barnett GROMACS教程3: 水中的多个甲烷
在本教程中, 我将向您展示如何创建一个包含多个OPLS甲烷的系统, 将其置于TIP4PEW水盒子中, 并根据模拟所得信息计甲烷-甲烷之间的平均力势.
设置
重复使用上一篇教程中的methane.pdb和topol.top(制作副本). 删除[ molecules ]下包含SOL的行, 这样就删除了topol.top中的所有水分子. 我们将模拟1000个水分子中的10个甲烷. 开始时, 我们将每个方向的盒子长度设置为3.2 nm. 显然, 当我们添加压力耦合后, 盒子会自动调整大小, 但我们希望初始的盒子大小接近平衡大小. 水的密度接近33.5分子/nm^3, 因此1010/33.5的立方根约为3.11, 向上舍入为3.2 nm.
首先, 将10个甲烷插入3.2 nm x 3.2 nm x 3.2 nm的盒子中:
$ gmx insert-molecules -ci methane.pdb -o box.gro -nmol 10 -box 3.2 3.2 3.2修改topol.top中指定甲烷数量的行, 将数字从1改为10.
然后往盒子里加水:
$ gmx solvate -cs tip4p -cp box.gro -o conf.gro -p topol.top -maxsol 1000gmx solvate会自动更新topol.top.
重复使用上次的.mdp文件, 但增大prd.mdp中的nsteps. 我自己运行了100 ns, 所以我只是在上次的数字末尾加了一个0. 您也可以提高输出频繁, 以便我们进行下面的分析.
模拟
像上次一样运行模拟.
分析
创建一个索引文件, 其中包含10个甲烷的分组:
$ gmx make_ndx -f conf.gro
> a C
> q现在使用gmx rdf, 就像上次一样, 但是选择包含C的组两次.
依赖于你保存了多少帧, 运行了多长时间的模拟(我运行了100 ns, 保存了25k帧), 如果只是简单地绘制RDF, 得到的结果应该如下所示:
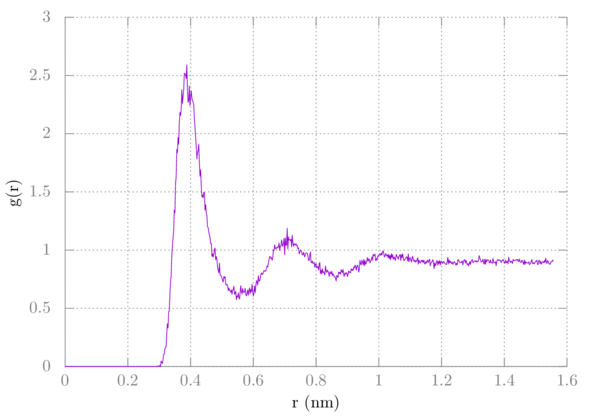
请注意, 它没有收敛到1. 我们多算了一个, 所以我们应该对其进行修正. 下面是gnuplot命令:
> plot 'rdf.xvg' u 1:($2*10/9) w l我们有10个甲烷, 但计算RDF时只用了9个, 所以我们将g的值乘以N/(N-1), 即10/9. 它应该如下所示:
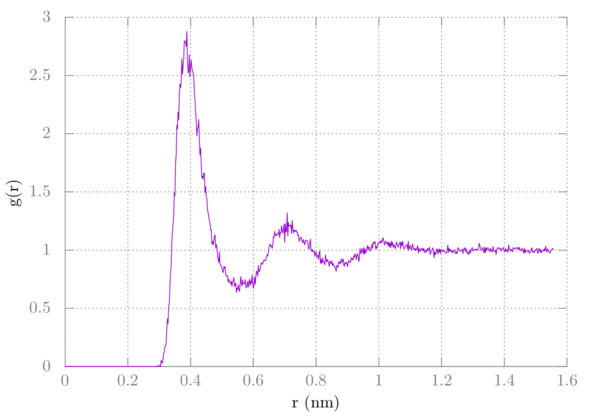
现在, 要从甲烷-甲烷RDF中得到甲烷-甲烷的平均力势(PMF), 我们要计算 $w = -kT\ln(g)$.
因此, 要用gnuplot绘图, 请执行以下操作:
> plot 'rdf.xvg' u 1:(-8.314e-3*298.15*log($2*10/9)) w l得到的结果应该类似下面:
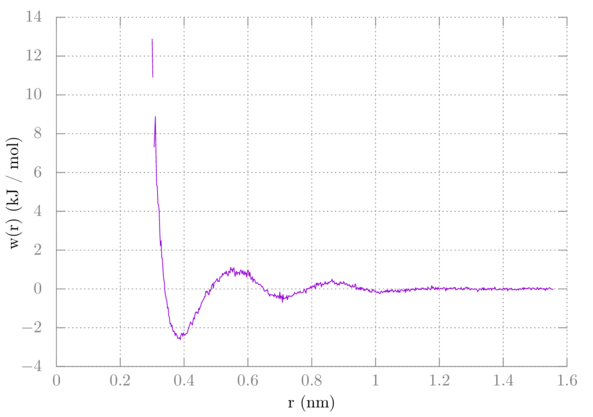
您注意到我们的抽样存在一个小的空白区. 这意味着, 在模拟中甲烷没有在那个距离有过相互作用. 我们将在后面的教程中使用伞形采样来解决这个问题.
总结
在本教程中, 我们使用gmx insert-molecules和gmx solvate创建了一个包含10个甲烷和1000个水的盒子. 我们使用上次的流程对其进行了模拟, 只不过模拟时间更长一点. 我们再次使用gmx rdf计算了径向分布函数, 但这一次算的是甲烷-甲烷之间的RDF. 由于这个原因, 我们不得不对结果进行修正, 根据修正后的RDF我们可以得到平均力势.
Barnett GROMACS教程4: 甲烷的溶剂化自由能
在本教程中, 我将向您展示如何使用GROMACS执行溶剂化自由能模拟, 以及如何使用MBAR计算自由能变化. 与往常一样, 本教程以之前的教程为基础, 特别是教程1和2.
如果您不熟悉自由能计算, 我建议您访问Alchemistry网站. 此页面与我们的教程尤其相关. 本教程将直接计算自由能. 对我们来说, 状态A是在水中完全相互作用的甲烷. 状态B将是一个甲烷, 它与水的所有范德华相互作用和库仑相互作用都被关闭; 甲烷可以与自身相互作用, 水可以与自身相互作用, 但甲烷和水不会相互作用. 就好像我们将甲烷从水中取出并置于真空中, 而在其他地方我们仍然拥有甲烷所在的水. 我们总共有15个状态, 我们将线性地关闭静电相互作用, 然后使用软核势来关闭范德华相互作用.
设置
创建模拟盒子
重复使用我们迄今为止用于OPLS甲烷和TIP4PEW水的methane.pdb和topol.top文件. 从topol.top文件中删除所有溶剂(删除以SOL开头的最后一行). 确保[ moleculetype ]以及[ molecules ]下的甲烷名称为Methane. 我们将使用一个甲烷, 因此如果有多个甲烷, 请将其更改为1.
这次我们使用不同的盒子类型, 这样我们使用的水可以少一点. 我们将使用距离甲烷每个方向1.2 nm的十二面体盒子. 首先创建这个盒子:
$ gmx editconf -f methane.pdb -bt dodec -d 1.2 -o box.gro现在填充溶剂:
$ gmx solvate -cs tip4p -cp box.gro -o conf.gro -p topol.top参数文件
我们将使用的参数文件与之前教程中的几乎完全相同, 只是我们添加了一个自由能部分, 以便慢慢关闭甲烷与水的相互作用. 另外, 我们还需要每个状态的参数文件. 我们需要15个最小化, 15个预平衡, 等等的参数文件. 但是我们将使用脚本来简单地更新参数文件模板中的适当值, 因此实际上只需要为模拟的每个部分提供一个模板. 在每个状态下, 我们将进行两次能量最小化, NVT预平衡100 ps, NPT预平衡1 ns, 然后进行5 ns的成品模拟.
这些文件可以这里下载.
以下是对其中一些新选项的解释说明:
| 参数 | 值 | 说明 |
|---|---|---|
| free-energy | yes | 进行自由能模拟 |
| init-lambda-state | MYLAMBDA | 本文件中的值实际上并不是我们运行模拟时使用的值. 它只是一个整数的占位符. 我们将模拟15个不同的状态, 因此这个数字的范围是 0到14.我们的脚本会将其替换为每个状态相应的值 |
| calc-lambda-neighbors | -1 | 输出每个状态的dH值, MBAR的必需选项 |
| vdw-lambdas | 见文件 | 关闭后静电, 我们以0.1的间隔关闭VDW作用.init-lambda-state = 0对应于此数组中的第一个值, init-lambda-state = 1对应第二个, 依此类推 |
| coul-lambdas | 见文件 | 关闭静电作用的步长为0.25. 类似vdw-lambdas, init-lambda-state指示使用哪一列 |
| couple-moltype | Methane | 要耦合/解耦的分子, 与拓扑文件中[ moleculetype ]的名称匹配 |
| couple-lambda0 | vdw-q | 当lambda为0(状态A)时, VDW和静电完全打开 |
| couple-lambda1 | none | 当lambda为1(状态B)时, 没有非键相互作用 |
| couple-intramol | no | 不关闭分子内的相互作用. 通常需要如此设置, 这样你就不必再重复真空中的模拟了 |
| nstdhdl | 100 | dH/dlambda的输出频率 |
| sc-alpha | 0.5 | 对VDW使用软核势. 该参数是软核函数中的一项. 请参阅手册 |
| sc-power | 1 | 如上 |
| sc-sigma | 0.3 | 如上 |
| sc-coul | no | 对静电作用不使用软核势 |
关于参数文件的最后一点说明: 我们使用sd积分器, 它代表随机动力学. sd本身可以控制温度, 所以我们不再使用Nose-Hoover热浴. 如果你使用md, GROMACS会给出警告, 提示你可能没有正确地对解耦状态进行采样.
模拟
如前所述, 我们实际上运行了15次模拟. 为了简化这一过程并避免使用15个不同的mdp文件作为输入, 请使用以下bash脚本来循环运行每个状态:
| 4-run.bsh | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 | #!/bin/bash
set -e
for ((i=0;i<15;i++)); do
sed 's/MYLAMBDA/'$i'/g' mdp/min.mdp > grompp.mdp
if [[ $i -eq 0 ]]; then
gmx grompp -o min.$i -pp min.$i -po min.$i
else
gmx grompp -c prd.$(($i-1)).gro -o min.$i -pp min.$i -po min.$i
fi
gmx mdrun -deffnm min.$i
sed 's/MYLAMBDA/'$i'/g' mdp/min2.mdp > grompp.mdp
gmx grompp -o min2.$i -c min.$i -t min.$i -pp min2.$i -po min2.$i -maxwarn 1
gmx mdrun -deffnm min2.$i
sed 's/MYLAMBDA/'$i'/g' mdp/eql.mdp > grompp.mdp
gmx grompp -o eql.$i -c min2.$i -t min2.$i -pp eql.$i -po eql.$i
gmx mdrun -deffnm eql.$i
sed 's/MYLAMBDA/'$i'/g' mdp/eql2.mdp > grompp.mdp
gmx grompp -o eql2.$i -c eql.$i -t eql.$i -pp eql2.$i -po eql2.$i
gmx mdrun -deffnm eql2.$i
sed 's/MYLAMBDA/'$i'/g' mdp/prd.mdp > grompp.mdp
gmx grompp -o prd.$i -c eql2.$i -t eql2.$i -pp prd.$i -po prd.$i
gmx mdrun -deffnm prd.$i
done |
该脚本使用了从i = 0到i = 14的for循环. 在每次迭代中, 它使用sed来查找我在mdp文件模板中放置的关键字MYLAMBDA, 并将其替换为正确的lambda状态. 然后此文件保存为grompp.mdp, 这是gmx grompp使用的默认文件名. 我们的所有输出都以lambda状态为后缀. 此外, 所有大于0的lambda状态都使用前一个状态的最终结构文件. 这对于甲烷的解耦并非完全必要, 但对其他系统可能会有帮助.
将上面的内容放到名为run.bsh的脚本中. 该脚本假定您已下载并将mdp文件放在名为mdp的子目录中. 然后执行:
$ chmod +x run
$ ./run分析
我们将使用MBAR, 具体来说就是称为pymbar的python实现. alchemical analysis脚本提供了一种简单的方法来执行计算, 实际上提供了几种不同的方法, 并可以进行误差分析.
下载并安装脚本后, 在包含结果的目录中运行它:
$ alchemical_analysis -p prd. -u kcal您的输出应如下所示:
States TI (kcal/mol) TI-CUBIC (kcal/mol) DEXP (kcal/mol) IEXP (kcal/mol) BAR (kcal/mol) MBAR (kcal/mol)
0 -- 1 0.002 +- 0.000 0.002 +- 0.000 0.002 +- 0.000 0.001 +- 0.000 0.002 +- 0.000 0.002 +- 0.000
1 -- 2 -0.000 +- 0.000 -0.000 +- 0.000 -0.001 +- 0.000 -0.000 +- 0.000 -0.000 +- 0.000 -0.001 +- 0.000
2 -- 3 -0.003 +- 0.000 -0.003 +- 0.000 -0.002 +- 0.000 -0.003 +- 0.000 -0.003 +- 0.000 -0.003 +- 0.000
3 -- 4 -0.005 +- 0.000 -0.005 +- 0.000 -0.005 +- 0.000 -0.006 +- 0.000 -0.005 +- 0.000 -0.005 +- 0.000
4 -- 5 0.093 +- 0.002 0.094 +- 0.002 0.090 +- 0.005 0.096 +- 0.003 0.095 +- 0.002 0.094 +- 0.001
5 -- 6 0.063 +- 0.002 0.065 +- 0.003 0.055 +- 0.010 0.063 +- 0.003 0.064 +- 0.002 0.060 +- 0.001
6 -- 7 0.013 +- 0.003 0.014 +- 0.003 0.025 +- 0.006 0.009 +- 0.003 0.014 +- 0.003 0.014 +- 0.002
7 -- 8 -0.055 +- 0.003 -0.052 +- 0.004 -0.051 +- 0.008 -0.050 +- 0.004 -0.052 +- 0.003 -0.052 +- 0.002
8 -- 9 -0.157 +- 0.004 -0.151 +- 0.005 -0.149 +- 0.014 -0.155 +- 0.006 -0.153 +- 0.004 -0.156 +- 0.003
9 -- 10 -0.338 +- 0.006 -0.324 +- 0.006 -0.306 +- 0.020 -0.332 +- 0.007 -0.330 +- 0.005 -0.336 +- 0.004
10 -- 11 -0.602 +- 0.005 -0.621 +- 0.006 -0.595 +- 0.016 -0.631 +- 0.006 -0.626 +- 0.005 -0.627 +- 0.004
11 -- 12 -0.674 +- 0.003 -0.708 +- 0.003 -0.708 +- 0.005 -0.709 +- 0.004 -0.708 +- 0.003 -0.711 +- 0.002
12 -- 13 -0.437 +- 0.001 -0.437 +- 0.002 -0.435 +- 0.002 -0.441 +- 0.002 -0.437 +- 0.001 -0.438 +- 0.001
13 -- 14 -0.138 +- 0.001 -0.134 +- 0.001 -0.131 +- 0.001 -0.132 +- 0.001 -0.131 +- 0.001 -0.130 +- 0.000
Coulomb: -0.007 +- 0.001 -0.007 +- 0.001 -0.006 +- 0.001 -0.007 +- 0.001 -0.007 +- 0.000 -0.007 +- 0.000
vdWaals: -2.232 +- 0.015 -2.255 +- 0.015 -2.205 +- 0.033 -2.281 +- 0.013 -2.265 +- 0.010 -2.282 +- 0.013
TOTAL: -2.238 +- 0.015 -2.262 +- 0.015 -2.211 +- 0.033 -2.288 +- 0.013 -2.272 +- 0.010 -2.289 +- 0.013
这是移除甲烷的自由能, 因此溶剂化自由能实际上是它的负数, 因为二者过程相反. 我们得到的结果2.289 kcal/mol与公布的数据相当. 在用TIP3P水模型模拟OPLS甲烷的论文中, 作者得到的结果为2.44 kcal/mol. 这两个结果的差距可能归因于使用了不同的水模型.
请注意, MBAR脚本给出了六种不同方法得到的不同结果. 其中一些方法比其他方法更好, 但所有六种方法都接近于对方, 告诉我们可能取样了足够多的状态来获得精确的数字. 其中一些方法比其他方法更好, 但所有六种方法彼此接近的事实意味着, 我们采样的状态可能足够多, 获得了精确的结果. 虽然BAR和TI也很好, 但MBAR是更好的方法之一. 我重新运行脚本以进行更多分析:
$ alchemical_analysis -p prd. -u kcal -g -f 20以下是每个lambda区间计算值的直观比较:
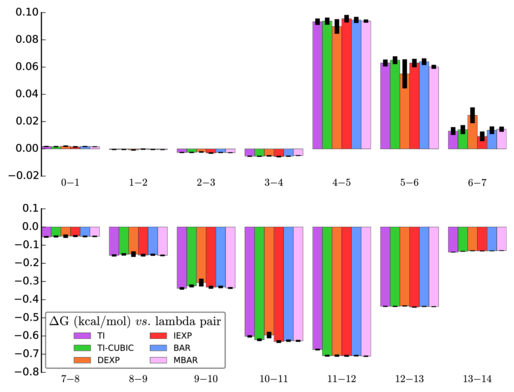
可以看到其中一种方法(DEXP)对状态5和6之间以及6和7之间的计算结果不同. 我并不太担心这个问题, 因为其他方法都符合得很好, 而且DEXP并不是”更好”的方法之一.
这是<dHdl>曲线:
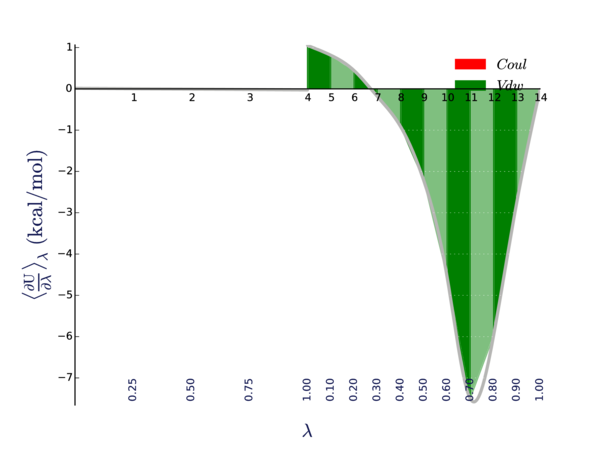
从这个图中, 我们可以看出曲率高的地方. 状态10, 11和12附近的曲率较高. 根据我们的应用, 我们可能希望在该区域中进行更多的采样.
最后, 这里是计算结果对时间的函数图, 既有前向模拟又后向模拟:
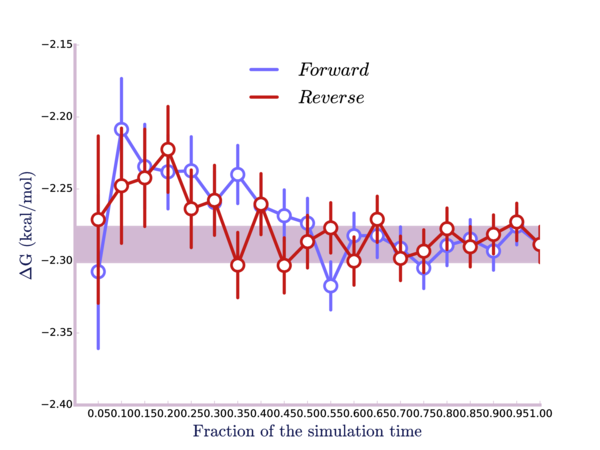
这里的要点是我们希望确保模拟平衡得足够好; 否则, 结果将不正确. 对于图第一部分中的任何重要部分, 如果曲线是平坦的, 就表明在开始模拟之前系统可能没有平衡好. 我们应该丢弃非平衡的数据并重新计算.
alchemical analysis脚本还支持一些其他命令行选项. 请务必查看脚本的主页和关于自由能分析最佳实践的论文, 其中详细介绍了这些内容以及和其他图形. 我不会在这里详细介绍所有选项.
总结
在本教程中, 我们对水中的甲烷进行了自由能模拟. 我们首先线性地关闭静电相互作用, 然后我们使用软核势关闭了范德华相互作用. 甲烷的分子内相互作用仍然存在, 因此就像我们从水中移除甲烷并将其置于真空中一样. 我们得到的结果2.289 kcal/mol与文献的结果相当.
Barnett GROMACS教程5: 使用伞形采样计算甲烷-甲烷的平均力势PMF
在本教程中, 我们将使用窗口采样, 有时也称为伞状采样, 来计算甲烷-甲烷的平均力势PMF. 在教程3中, 我们通过模拟几个甲烷分子计算径向分布函数的方法直接获得了PMF. 像这样对几个溶质分子进行采样的方法并不总是可行的.
我们要用两个甲烷分子, 并使用简谐势将其约束在不同距离. 不同的距离称为”窗口”. 在常规模拟中, 就像我们在教程3中所做的那样, 其中一些窗口很难采样. 伞形采样通过迫使分子保持在设定距离的一定范围内来解决这个问题.
最后, 我们将使用GROMACS实现的加权直方图分析(WHAM)来重建PMF. Alchemistry维基上的一篇文章讨论了存在化学变化情况下的WHAM. 在这里, 我们的反应坐标是两个甲烷之间的距离.
设置
创建盒子
我们将再次使用之前教程中的methane.pdb和topol.top. 使用gmx insert-molecules将两个甲烷插入盒子中, 然后使用gmx solvate溶剂化盒子. 在本教程中, 需要使用立方体盒子, 边长至少为3.1 nm.
创建索引文件
我们还需要创建一个索引文件, 其中包含我们感兴趣的两个组, 它们之间受伞形势限制. 使用gmx make_ndx创建一个索引文件, 其中包含每个甲基碳的组, 并将其分别命名为CA和CB.
$ gmx make_ndx -f conf.gro然后, 假定残基CH4在第2组中:
> splitres 2现在假定最后两组是6和7, 它们是两个甲烷分子:
> 6 & a C
> 7 & a C现在命名这些组:
> name 8 CA
> name 9 CB
> q使用gmx make_ndx的其他方法同样可以达到目的. 关键是, 需要将每个甲烷的碳原子放在单独的索引组中, 并分别将它们命名为CA和CB.
参数文件
我们在很大程度上重复使用前几个教程中的参数文件, 但是我们将添加一个与质心(COM)牵引有关的部分. 牵引代码可以将甲烷维持在指定距离. 可能有几种不同的设置方法, 但对于这个系统, 我们将手动指定两个甲烷间的距离.
每个步骤的参数文件都可以在这里下载.
就像在自由能教程中一样, 这些文件是带有关键字的模板, 这些关键字将使用bash脚本替换. 这是因为我们必须为每个窗口运行一套完整的模拟, 并需要指定每个窗口的距离.
以下是对文件中使用的新参数的解释说明:
| 参数 | 值 | 解释说明 |
|---|---|---|
| pull | yes | 使用牵引代码 |
| pull-ngroups | 2 | 有两个牵引组 |
| pull-group1-name | CA | 索引文件中指定的组名称. 对我们来说, 这是其中一个甲烷的碳, 尽管我们也可以选择整个甲烷. 如果那样做的话, 就会沿着两个甲烷分子的质心方向进行牵引 |
| pull-group2-name | CB | 另一个甲烷的碳原子 |
| pull-ncoords | 1 | 只沿一个坐标方向进行牵引 |
| pull-coord1-geometry | distance | 沿两个组连线的方向进行牵引 |
| pull-coord1-type | umbrella | 对此牵引坐标使用伞形(简谐)势 |
| pull-coord1-groups | 1 2 | 对此坐标牵引的两个组(定义见后面). 实际上你可以定义更多的牵引坐标, 也可以在不同的分子组之间进行牵引, 但我们在这里不使用这些选项 |
| pull-coord1-k | 5000.0 | 伞形势的力常数, 单位kJ/(mol-nm) |
| pull-coord1-init | WINDOW | 两个组的初始间距. 这是关键字, 我将使用bash脚本将其替换为每个窗口的相应值 |
| pull-coord1-rate | 0.0 | 我们不需要两个组沿坐标方向运动, 因此设置为零 |
| pull-coord1-start | no | 我们手动指定每个窗口的距离, 因此计算时不需要考虑质心距离 |
参数文件设置为100 ps NVT预平衡, 然后是1 ns NPT预平衡, 最后是5 ns成品模拟. 我们希望在平衡过程中施加伞势时, 甲烷能维持正确的距离. 对于其他一些系统, 您可能必须使用更合适的方法每个窗口生成初始构型.
模拟
对于模拟, 我们将使用bash脚本替换mdp文件中的WINDOW关键字, 非常类似于我们在自由能模拟中的做法. 以下是脚本:
| 5-run.bsh | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 | #!/bin/bash
set -e
for ((i=0;i<27;i++)); do
x=$(echo "0.05*$(($i+1))" | bc);
sed 's/WINDOW/'$x'/g' mdp/min.mdp > grompp.mdp
gmx grompp -o min.$i -pp min.$i -po min.$i -n index.ndx
gmx mdrun -deffnm min.$i -pf pullf-min.$i -px pullx-min.$i
sed 's/WINDOW/'$x'/g' mdp/min2.mdp > grompp.mdp
gmx grompp -o min2.$i -c min.$i -t min.$i -pp min2.$i -po min2.$i -maxwarn 1 -n index.ndx
gmx mdrun -deffnm min2.$i -pf pullf-min2.$i -px pullx-min2.$i
sed 's/WINDOW/'$x'/g' mdp/eql.mdp > grompp.mdp
gmx grompp -o eql.$i -c min2.$i -t min2.$i -pp eql.$i -po eql.$i -n index.ndx
gmx mdrun -deffnm eql.$i -pf pullf-eql.$i -px pullx-eql.$i
sed 's/WINDOW/'$x'/g' mdp/eql2.mdp > grompp.mdp
gmx grompp -o eql2.$i -c eql.$i -t eql.$i -pp eql2.$i -po eql2.$i -n index.ndx
gmx mdrun -deffnm eql2.$i -pf pullf-eql2.$i -px pullx-eql2.$i
sed 's/WINDOW/'$x'/g' mdp/prd.mdp > grompp.mdp
gmx grompp -o prd.$i -c eql2.$i -t eql2.$i -pp prd.$i -po prd.$i -n index.ndx
gmx mdrun -deffnm prd.$i -pf pullf-prd.$i -px pullx-prd.$i
done |
我们模拟了26个窗口, 距离从0.05 nm到大约1.3 nm. 请注意, 我为每次模拟都添加了牵引力输出文件选项-pf和牵引距离输出文件选项-px. 这是因为使用-deffnm选项时GROMACS会覆盖这两个文件. 此外, 我使用-n指定了索引文件, 因为gmx grompp需要获取我们使用牵引参数指定的组. 请注意, 我使用bc的小技巧, 以便在bash中使用浮点数进行数学运算.
分析
我们将使用gmx wham来获取PMF. 该程序接受一个包含.tpr文件列表的文件和另一个包含.xvg文件列表的文件, 后者中包含了输出的牵引力.
要创建这两个文件, 请执行以下操作:
$ ls prd.*.tpr > tpr.dat
$ ls pullf-prd.*.xvg > pullf.dat然后你可以运行gmx wham:
$ gmx wham -it tpr.dat -f pullf.dat运行gmx wham之后, 平均力势的计算结果保存在名为profile.xvg的文件中. 绘图应该如下所示:
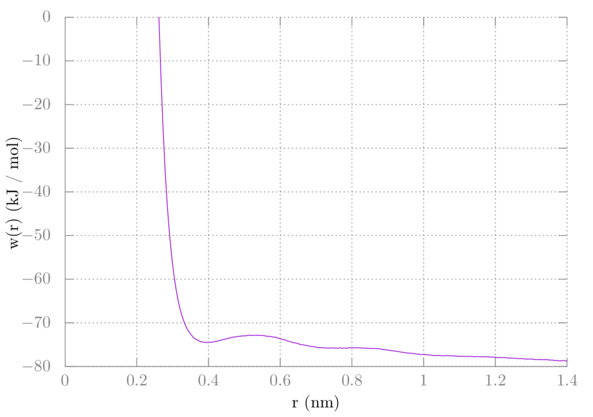
我们希望在较远的距离处相互作用为零. 但是, 由于我们使用了三维的偏置势, 需要进行校正. 想象一下将其中一个甲烷作为参考点. 另一个甲烷可以在该点周围距离 $r$ 处进行采样, 覆盖了半径为 $r$ 的某个球体的表面. 这为我们的采样增加了额外的构型空间, 降低了熵. 需要消除这些额外熵对PMF的贡献. 回想一下, 等温等压系综中的吉布斯自由能是 $-kT\ln(W)$, 其中 $W$ 为分配函数. 对我们的情况, 甲烷绕着球体表面采样时, $W$ 与该球体的表面积成正比. 因此, 需要增加 $2kT\ln(r)$ 的校正. 此外, 我们需要将图形向上移动, 使其尾部变为零. 对我的结果需要增加的数值大约为77, 但你需要增加的数值可能不同. 要在gnuplot中绘制此图, 请在gnuplot终端中执行以下操作:
> plot 'profile.xvg' u 1:($2+2*8.314e-3*298.15*log($1)+77) w l您的PMF现在应该如下所示:
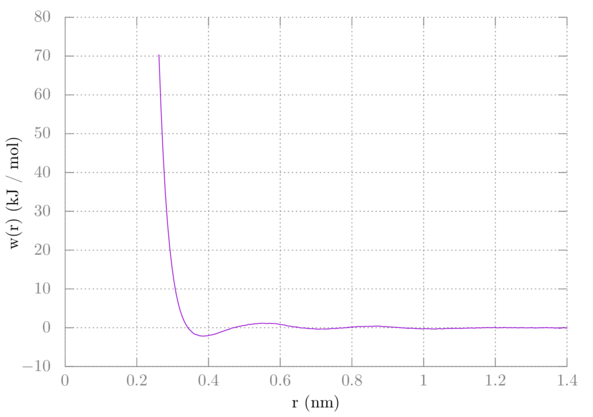
与教程3中的PMF相比, 可以看到它们几乎相同:
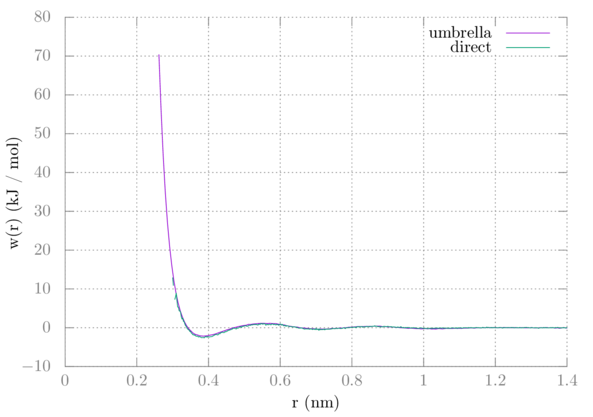
区别在于, 使用教程3的直接方法, 采样不可能接近窗口采样. 两个甲烷不会自然地相互靠近, 这就是为什么我们必须增加伞形势以维持它们之间距离的原因.
另一个输出是histo.xvg, 它有助于确定窗口之间是否有足够的重叠. 以下是我模拟的每个直方图的绘图:
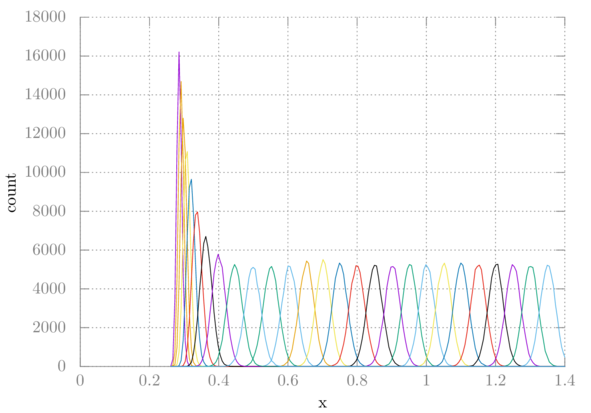
很显然, 我们的窗口已经足够重叠了. 否则的话, 我们可能必须选择较小的窗口, 或选择缺少模拟的特定点进行模拟.
总结
在本教程中, 我们使用GROMACS质心牵引代码对水中的两个甲烷进行窗口采样. 然后我们使用gmx wham计算了平均力势.
Barnett GROMACS教程6: 使用测试粒子插入计算甲烷的超额化学势
在本教程中, 我们将使用测试粒子插入(TPI)来计算甲烷溶解在水中的超额化学势. 大多数用户都不知道GROMACS有一个用于运行TPI的内置方法. 本教程不会全面讨论TPI的统计力学原理, 但在需要时会进行说明. 鼓励用户寻找有关此方法的科学资料.
TPI涉及将某些状态微扰到其他非常相似的状态. 我们将采用块体水并插入一个甲烷粒子, 测量由此产生的势能变化. 势能的这一变化与超额化学势之间存在统计学上的关系. 对我们来说, 状态A是块体水系统, 状态B是含有甲烷的水系统.
您需要使用GROMACS对状态A进行普通的MD模拟. 我们已经在教程1中对块体水进行了此模拟. 我们将重复使用那里得到的输出轨迹文件来插入甲烷.
设置
创建水系统
按照教程1运行包含TIP4PEW水的系统.
将测试粒子添加到拓扑文件
我们原来的拓扑文件中只有水. 在新的拓扑文件中, 我们只需要添加1个测试粒子, 它必须是系统中的最后一个分子. 我们将使用’opls_066`作为粒子的原子类型, 这是OPLS联合原子甲烷. 这是我的最终拓扑文件(对你的系统, 水的数目可能不同):
| topol.top | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 | #include "oplsaa.ff/forcefield.itp"
#include "oplsaa.ff/tip4pew.itp"
[ moleculetype ]
; Name nrexcl
Methane 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass
1 opls_066 1 CH4 C 1 0 16.043
[ System ]
Methane in water
[ Molecules ]
SOL 395
Methane 1 |
将测试粒子添加到gro文件
您还需要将测试粒子添加到gro文件中. 只需编辑conf.gro(或使用的任何其他.gro文件), 并在末尾添加一行包含测试粒子位置的文本(在盒子坐标之前). 我添加的一行如下所示:
396CH4 C 1581 0.000 0.000 0.000
实际位置无关紧要; GROMACS只需要测试粒子的占位符. 此外, 您还需要将.gro文件第二行中系统的粒子总数增加1.
参数文件
我们只需要一个TPI参数文件. 只需复制块体水模拟中的prd.mdp并将integrator更改为tpi. 您应该将nsteps更改为每帧中想要尝试的插入次数. 我选择了100000次. 您还需要将cutoff-scheme更改为group, 因为Verlet尚未支持TPI.
模拟
对于模拟, 我们只是使用保存的轨迹文件(在第一个教程中命名为prd.xtc)重新运行块体水模拟. 要做到这一点, 首先运行grompp:
$ gmx grompp -f mdp/tpi.mdp -o tpi -po tpi -pp tpi -c conf.gro现在使用-rerun选项运行mdrun:
$ gmx mdrun -deffnm tpi -rerun prd.xtc分析
日志文件(在本例中名称为tpi.log)中包含了平均体积和平均差额化学势. 我得到的结果如下:
<V> = 1.18704e+01 nm^3
<mu> = 8.81230e+00 kJ/mol
<mu>的单位为kJ/mol, 如果将其转换为kcal/mol, 我们得到2.106 kcal/mol. 这与我们在教程4中使用λ耦合方法得到的溶剂化自由能2.289 kcal/mol一致. 结果的差距可以归因于模拟溶剂化自由能时使用了全原子模型, 而在本教程中使用了联合原子模型.
运行结束后还会给出`prd.xvg’文件, 里面包含了运行过程中各种能量的信息
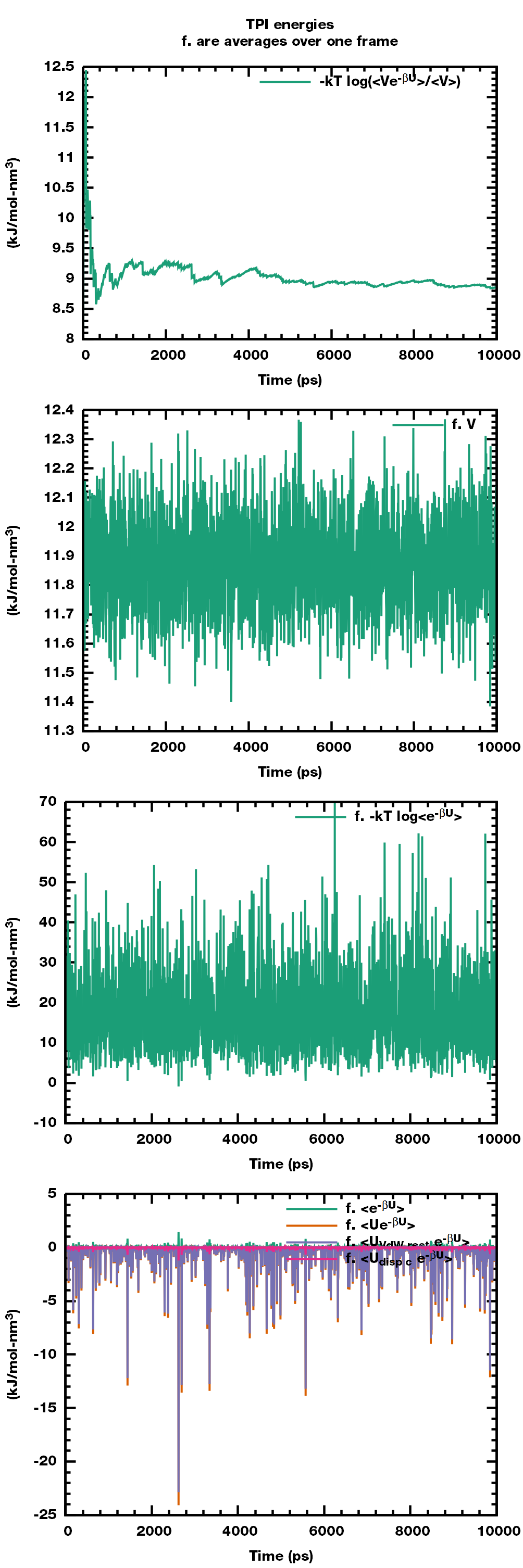
tpi.xvg文件中是TPI能量的分布
总结
在本教程中, 我们示例了如何使用GROMACS进行测试粒子插入模拟, 以获得OPLS联合原子甲烷的超额化学势.
