- 2016-03-11 21:18:39 初稿
- 2016-03-22 14:28:06 补充 程刚 的方法
- 2016-03-27 22:02:21 补注
理论考虑
分子动力学模拟的分析是动力学的精髓部分, 要依据模拟的假设和目的去分析所得到的数据, 看是否支持自己的假设, 进而得到更多的洞察. 没有假设和目的就去盲目地做模拟, 是不可取的. 由于各人所作体系不同, 对于分析没有通用的套路, 你需要多参考文献才好.
虽然GROMACS自带了很多分析工具, 可能仍不能满足你的需要, 这时候就需要你自己写代码对轨迹进行分析了. 对此, 我的建议如下:
- 如果可能, 请尽量使用GROMACS自带的工具完成分析过程, 必要时可以组合多个分析工具达到目的. 虽然这种做法处理时间可能稍长, 但无须写代码, 适合普通用户使用. 但这种做法需要你对GROMACS自带的工具有比较详尽的了解, 知晓各个工具的功能, 并能根据自己的目的进行恰当地组合.
- 即便需要自己写代码分析轨迹, 仍然建议先利用GROMACS自带的工具对轨迹进行初步处理, 如利用
gmx trjconv转换轨迹格式, 处理居中, PBC问题; 利用gmx select获取特定原子的索引; 利用gmx trjconv或gmx traj抽取特定索引原子的信息等. 因为无论做哪种分析, 都要牵涉到这些问题的处理, 而自己写代码实现这些功能虽然不是很可能, 但相当麻烦, 而且执行效率也没有使用GROMACS自带工具高. 这样得到需要的原子坐标后, 我们就可以把注意力集中到自己需要分析方面, 节省了时间和精力. - 如果为了提高处理轨迹的效率或其他原因, 你不得不自行处理轨迹, 建议你使用
xtc格式. 这种压缩格式只包含坐标, 文件小, 当只需要分析坐标时, 使用它可大大提高效率. 但如果你同时需要坐标和速度甚至受力, 那你就只能使用trr格式了. - 具体如何使用C, Fortran或MatLab来处理
xtc或trr格式的轨迹, 请参考博文. - 如果熟悉, VMD的tcl脚本也可用于分析轨迹, 缺点是速度慢, 且对文件大小有限制.
具体示例
下面我们模拟TIP3P水中的一个甲烷CH4分子, 以计算CH4第一溶剂化层中水分子的平均滞留时间为例, 来对分析过程进行具体地说明.
下载示例文件, 解压后得4个文件, conf.gro, grompp.mdp, index.ndx, topol.top. 值得注意的是, index.ndx文件使用gmx make_ndx添加了每种原子的索引号, 这是为了便于后面的分析.
体系中包含一个CH4分子(OPLS-AA力场)和241个TIP3P水分子
下面我们来运行模拟
grompp -maxwarn 2
mdrun
作为示例, 我们只运行了20 ps, 得到了轨迹文件traj.xtc.
要计算CH4分子第一溶剂化层中水分子的平均滞留时间, 我们首先需要知道第一溶剂化层的厚度, 为此, 我们可以计算CH4分子和水分子二者质心之间的径向分布函数RDF
g_rdf -f -n -rdf mol_com
> 2 3
得到如下图形
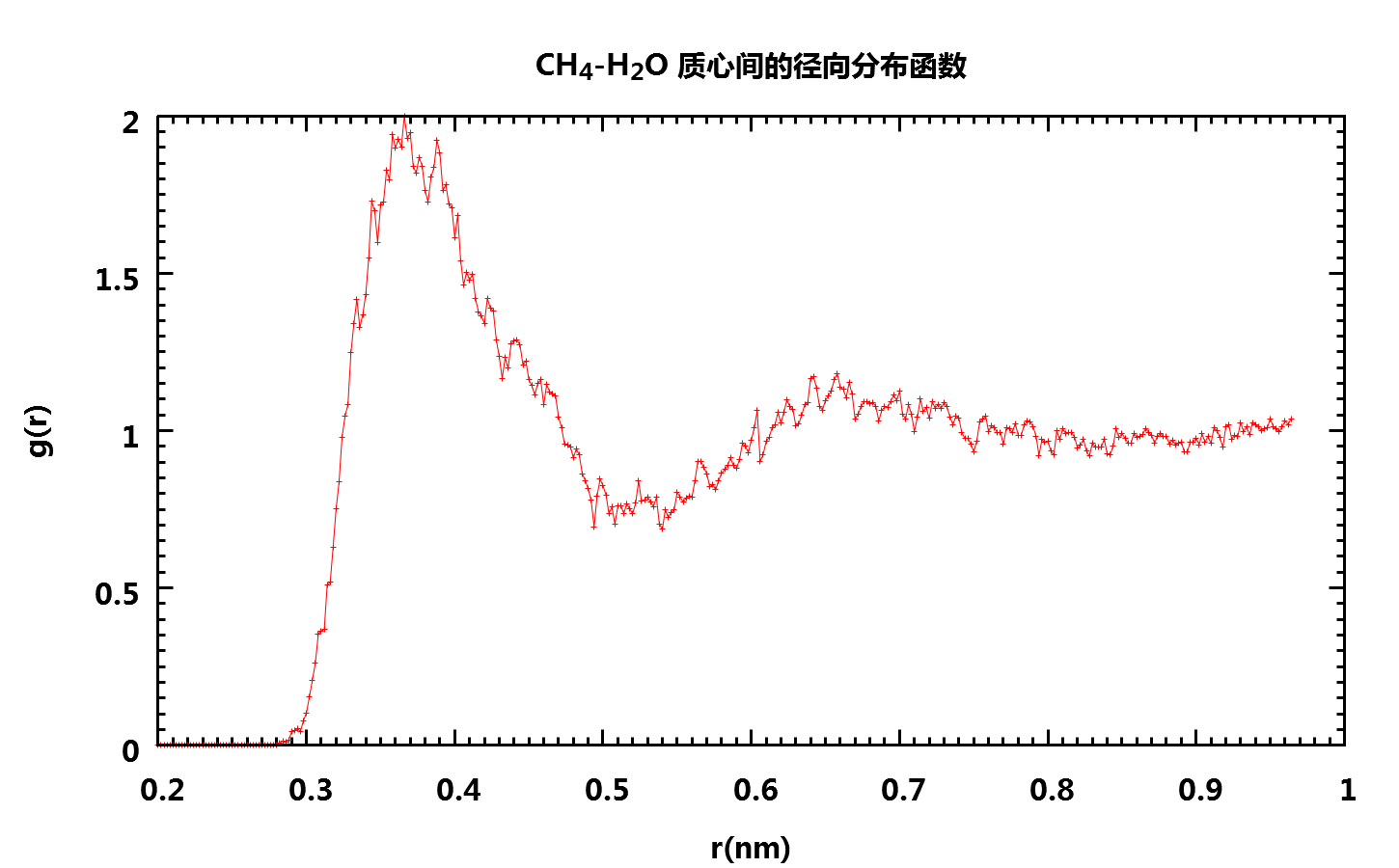
可以看到, 由于模拟时间较短, 得到的RDF并不是十分光滑(延长模拟时间就可以得到更漂亮的图形), 但我们仍然可以看出第一溶剂化层的厚度大约是0.5 nm. 也就是说, 我们在计算平均滞留时间时, 只考虑其质心处于CH4质心0.5 nm 范围内的水分子.
【2016-03-27 补注】上面关于第一溶剂化层厚度的说法不严谨. 文献上一般以RDF的第一个峰值作为第一溶剂化层的厚度, 所以根据上图应该是0.36 nm左右, 下面分析中应该使用这个数值.
我们使用g_select工具来获取每个水分子的是否处于CH4第一溶剂化层中的信息.
g_select -f -n -os -oc -oi -om -on selFrm.ndx -selrpos mol_com
>"1st shell" resname SOL and name OW and within 0.5 of resname CH4
在上面的命令中我们为-on选项指定了输出文件, 以防止默认的输出文件index.ndx与前面我们使用的index.ndx文件冲突. 有关g_select的使用说明请参考其说明文档和GROMACS选区(selection)语法及用法.
我们得到如下文件
size.xvg: 每一时刻第一溶剂化层中原子的个数cfrac.xvg: 每一时刻第一溶剂化层中原子的覆盖比例index.dat: 每一时刻第一溶剂化层中原子的个数及其编号mask.dat: 每一时刻分子是否处于第一溶剂化层中的掩码,0: 不处于,1:处于selFrm.ndx: 每一时刻第一溶剂化层中原子的索引组
请打开这些文件进行查看, 了解其含义.
我们只要对mask.dat进行分析处理就可以计算平均滞留时间了. 这可以使用下面的bash脚本完成
| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 | file=mask.dat
ftrs=${file%.*}_trs.dat
ffrq=${file%.*}_frq.dat
awk ' BEGIN {
getline
while($1=="#") getline
Ncol=NF
close(FILENAME)
for(i=2; i<=Ncol; i++) {
while(getline<FILENAME) if(NF==Ncol) printf "%s", $i
print ""
close(FILENAME)
}
}' $file > $ftrs
awk -v file=$file '
BEGIN{ Navg=0; Tavg=0
getline <file
while($1=="#") getline <file; dt=$1
getline <file; dt=$1-dt
close(file)
}
{ gsub(/0+/, " ")
Ntxt=split($0, txt)
for(i=1; i<=Ntxt; i++) {
T=length(txt[i])
print T
Navg++; Tavg += T
}
}
END{ print "# Avaraged Residence Time(ps)=", Tavg*dt/Navg}
' $ftrs >$ffrq
|
得到的平均滞留时间为0.807243 ps. 当然, 实际情况中你需要运行更长的模拟来确认得到的数据是否收敛. 水分子滞留时间的分布图如下
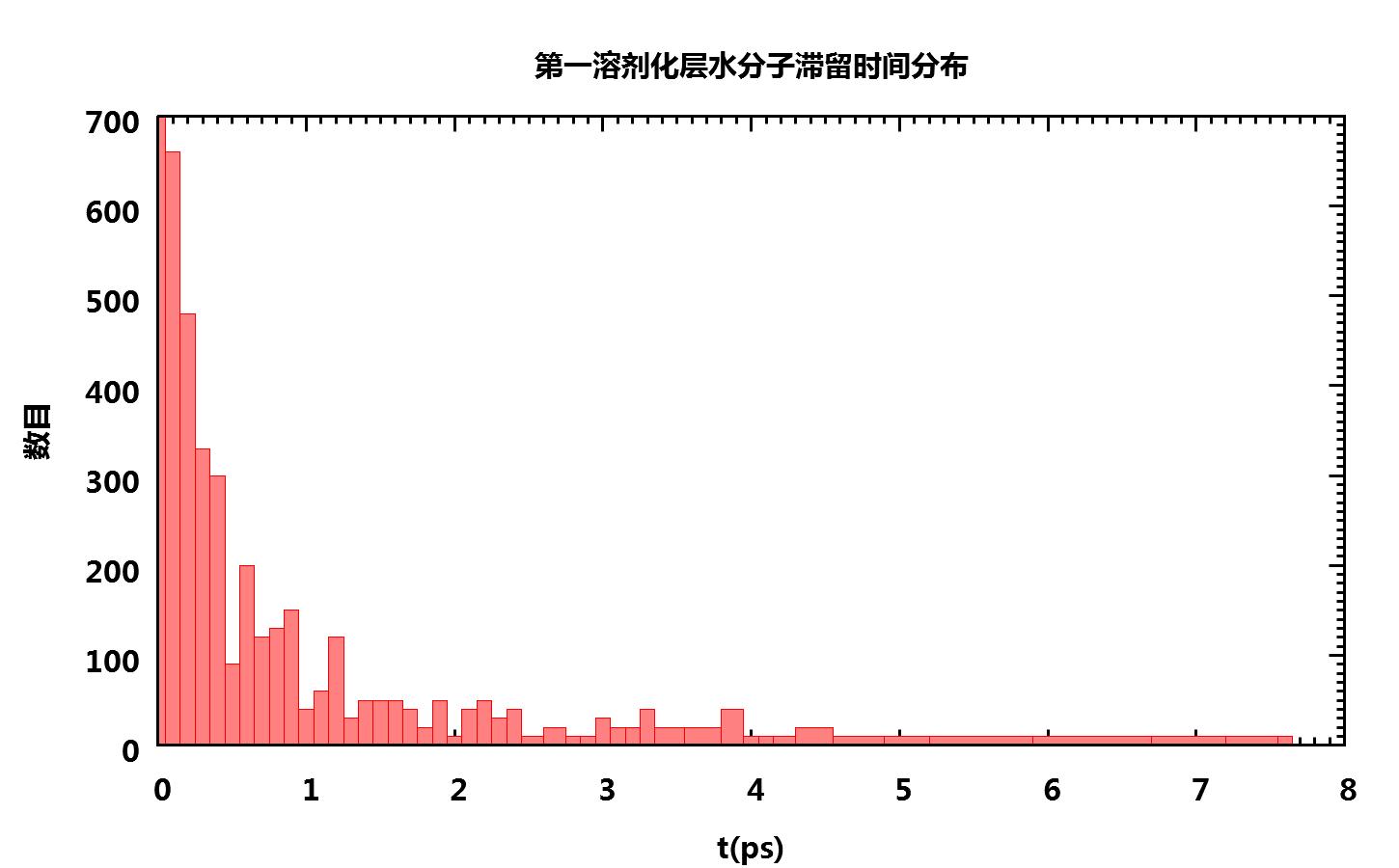
对更大的体系, 更长的模拟时间, 上面的简单脚本可能执行时间很长. 这主要是因为在第一步中对mask.dat进行行列互换时, 如果文件太大就要花费很长的时间. 一种更高效些的方法是使用中间文件, 方法如下
| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | awk -v ftrs=$ftrs ' BEGIN{ Nmax=2000; N=0
system("rm -rf _row-* " ftrs " && cat <> " ftrs)
}
$1!="#" {
N++
Frow="_row-"sprintf("%04d",N)
$1=""; sub(/^\s*/, ""); gsub(" ", "\n", $0)
print $0 >Frow
close(Frow)
if(N>Nmax) {
N=0
system("ls _row-* | sort | xargs paste -d \047\047 "ftrs" >_trsRow")
system("rm -rf _row-* && mv _trsRow "ftrs)
}
}
END{
system("ls _row-* | sort | xargs paste -d \047\047 "ftrs" >_trsRow")
system("rm -rf _row-* && mv _trsRow "ftrs)
}
' $file
|
更高效的方法, 就只能换用其他编译型语言或MatLab等软件了.
我们可以利用trjconv程序并借助selFrm.ndx文件获取每一时刻所选原子的坐标, 只需要根据对每一帧指定不同的索引组即可. 获取前100帧的示例代码如下
| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 | file=selFrm.gro
echo -n ""> $file
dt=0.002
for i in {0..100}; do
t=$(echo "$dt*$i" | bc)
echo $i | trjconv -f -n selFrm.ndx -dump $t -o _tmp.gro 2>/dev/null
cat _tmp.gro >>$file
rm -rf _tmp.gro
done
|
当然这种每次处理一帧的方法运行起来很慢, 但可惜的是GROMACS的分析工具中并没有提供解决方案, 如果需要更快地抽取出构型, 那就只能自己写代码了. 此外, 这样直接得到的构型由于PBC的原因可能看起来不连续, 为此, 你可能需要先使用trjconv对轨迹进行居中, PBC处理, 然后再使用上面的方法获取坐标.
网络资料
- 如何计算平均滞留时间(residence time)
- trajectory output from g_select?
- gmx dipoles with dynamic indices (gromacs 5.0.x)
- extract coordinates of selected atoms
- Windows Cmd终端Ctrl D不起作用的解决方法
- Equivalent to ^D (in bash) for cmd.exe?
- LINUX SHELL 行列转换、倒序
程刚: 基于GROMACS和MatLab计算平均滞留时间
一、计算原理
可参考文献 Phys. Chem. Chem. Phys., 2012, 14, 16536-16543
划定一定的区域,定义为系统计算空间,比如距离聚合物膜0.5 nm的空间。假设体系里有N个分子,每个分子在系统计算空间的平均滞留时间都不同,对体系的N个分子全部进行平均,就得到了平均滞留时间。下面以计算水分子的平均滞留时间为例。
假设统计的轨迹共n帧,共计x ns,统计轨迹时间x ns应该远大于水分子的滞留时间。以第1号水分子为例,统计这n帧轨迹中第1号水分子在系统计算空间内出现的总时间,再除以第1号水分子进出此空间的次数,就得到了第1号分子的平均滞留时间,记为T(1)。然后对所有的水分子的平均滞留时间进行平均,记为Tavg=sum(T)/n,其中sum为加和函数,n为出现在系统计算空间的总分子数(即排除所有未曾出现在系统计算空间内的分子)。
二、计算过程
1. 采用GROMACS软件对轨迹进行处理
g_select -f md.xtc -s md.tpr -n index.ndx -om M_1.dat -select 'name OW and within 0.195 of group M'
系统计算空间为距离M基团0.195 nm的空间;其中md.xtc文件为轨迹,共计n帧;index.ndx索引文件中应该提前设定好待分析的组和OW原子;产生的M_1.dat文件是二进制的二维数据表,第一列为轨迹时间,后面每列为每个OW原子在每个轨迹时间点上在系统计算空间内的出现情况(1代表出现,0代表不出现)。
2. 采用MatLab软件处理dat文件
使用下面的MatLab代码处理得到的M_1.dat文件,注意文件名要匹配。程序运行结束后,Tavg为所求。
李继存 注: 此代码效率较低, 数据量太大时可能出现问题, 请优化使用.
| matlab | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 | function ResTime
clc; clear all;
global Ntime Natom
dt=2; % 时间间隔
mat=load('SO3_1.dat') ; % mask数据文件
[Ntime,Ncol]=size(mat); %
mat=mat(1:Ntime,2:Ncol); %
Natom=Ncol-1;
T=zeros(1,Natom); % 某一列的滞留时间
Tnum=zeros(1,Natom); % 未连续出现的次数统计
Ttot=zeros(1,Natom); % 某一列的总和,出现的总时间
for i=1:Natom
Ttot(i)=sum(mat(:,i));
if(Ttot(i)~=0)
for j=1:Ntime-1
if mat(j+1,i)~=mat(j,i)
Tnum(i)=Tnum(i)+0.5;
end
end
T(i)=double(Ttot(i)/ceil(Tnum(i)));
end
end
Tavg=sum(T)*dt/length(find(Ttot>0))
|
评论
- 2016-10-20 11:40:17
wlzzl第一个脚本运行总是错误,是复制下来格式不对吗? - 2016-10-21 12:46:48
Jerkwin说清楚, 是哪个脚本, 错误提示是什么? - 2016-10-24 20:01:30
wlzzl就是第一个利用mask.dat求驻留时间的脚本,awk指令的最后那个单引号,没有跟大括号在同一行,运行时总是报错,也不知什么原因。 - 2016-10-25 15:10:25
Jerkwin系统的原因吧, 可能和换行符什么的有关 -
2016-10-24 19:45:37
wlzzl谢谢了,翻看了书,已经解决。 - 2016-11-02 15:29:48
live想问下,利用第一个脚本文件算出的滞留时间和文献差异大吗?我算出来差异有点大。 -
2016-11-02 21:12:52
Jerkwin这种方法有其本身的缺点, 计算得到的值和使用的时间步长有关. 更好的方法你可以参考文献. - 2017-02-26 16:07:46
Topin李老师,5.0+版Gromacs, gmx select里-olt项输出的lifetime.xvg文件就是滞留时间分布数据吧? - 2017-02-26 19:05:25
Jerkwin有人说是, 但是我没有测试过, 不清楚到底是不是. 你可以测试一下, 如果确定了, 请告诉我一声, 以便我更新下文章.
