- 2016-06-08 10:40:58 翻译整理: 李继存; 校对: 王浩博
【按】文章来源于Science上的一篇论文. 对于此文网上原有一翻译, 未知译者, 但尚嫌粗糙, 且舛误颇多. 我以其为底稿, 重新翻译整理后, 供大家参考.
The Protein-Folding Problem, 50 Years On
蛋白质折叠问题50年的历程
Abstract
摘要
The protein-folding problem was first posed about one half-century ago. The term refers to three broad questions: (i) What is the physical code by which an amino acid sequence dictates a protein's native structure? (ii) How can proteins fold so fast? (iii) Can we devise a computer algorithm to predict protein structures from their sequences? We review progress on these problems. In a few cases, computer simulations of the physical forces in chemically detailed models have now achieved the accurate folding of small proteins. We have learned that proteins fold rapidly because random thermal motions cause conformational changes leading energetically downhill toward the native structure, a principle that is captured in funnel-shaped energy landscapes. And thanks in part to the large Protein Data Bank of known structures, predicting protein structures is now far more successful than was thought possible in the early days. What began as three questions of basic science one half-century ago has now grown into the full-fledged research field of protein physical science. 蛋白质折叠问题大约半个世纪前第一次提出. 这一术语涉及三大问题: (i) 氨基酸序列决定蛋白质天然结构的物理规则是什么? (ii) 蛋白质怎么能折叠得如此迅速? (iii) 我们能不能设计一种计算机算法, 根据蛋白质的序列来预测其结构? 我们将对在这些问题上取得的进展进行综述. 在一些例子中, 通过计算化学细度模型中的物理作用力, 计算机模拟已经能够实现小型蛋白质的精确折叠. 我们已经知道蛋白质折叠得很迅速是因为随机热运动导致其构象发生变化, 致使蛋白质沿能量下降途径趋向其天然结构. 这一原理可由漏斗状的能量形貌进行说明. 部分地归功于蛋白质数据库(PDB, Protein Data Bank)中大量的已知结构, 现在预测蛋白质结构远比早期预想的要成功. 半个世纪前, 由这三个基础科学问题开始的研究现如今已经发展成为蛋白质物理科学研究的成熟领域. Protein molecules embody a remarkable relationship between structure and function at the molecular level. Proteins perform many different functions in biochemistry. A protein's biological mechanism is determined by its three-dimensional (3D) native structure, which in turn is encoded in its 1D string of amino acid monomers. 蛋白质分子在分子层面上体现了蛋白质结构与其功能之间的显著关系. 在生物化学中蛋白质有许多不同的功能. 一种蛋白质的生物学机制由其三维(3D)天然结构决定, 而蛋白质的天然结构又由其氨基酸单体的一维链进行编码. This year marks the 50th anniversary of the 1962 Nobel Prize in Chemistry awarded to Max Perutz and John Kendrew for their pioneering work in determining the structure of globular proteins (1-3). That work laid the foundation for structural biology, which interprets molecular-level biological mechanisms in terms of the structures of proteins and other biomolecules. Their work also raised the question of how protein structures are explained by physical principles. Upon seeing the structure of myoglobin (Fig. 1) at 6 Å resolution (1), Kendrew et al. said, 1962年的诺贝尔化学奖授予了Max Perutz和John Kendrew, 以表彰他们在确定球蛋白结构方面所做的开创性工作(1-3), 而今年是其50周年纪念. 他们的工作奠定了结构生物学的基础, 从蛋白质和其他生物分子的角度解释了分子层面上的生物学机制. 他们的工作也提出这样的问题, 如何用物理原理来阐明蛋白质的结构. 当看到6 Å分辨率下肌红蛋白的结构(图 1)时, Kendrewet等人指出, Fig. 1 In 1958, Kendrew and co-workers published the first structure of a globular protein: myoglobin at 6 Å resolution (1). Its puzzlingly complex structure lacked the expected symmetry and regularity and launched the protein-folding problem. [With permission from the Medical Research Council Laboratory of Molecular Biology]
图 1 1958年, Kendrew及其合作者发表了球状蛋白的第一个结构: 6 Å分辨率下的肌红蛋白(1). 它的结构莫名其妙地复杂, 并缺乏预想的对称性和规则性. 这一事实开启了蛋白质折叠问题的研究.
Fig. 1 In 1958, Kendrew and co-workers published the first structure of a globular protein: myoglobin at 6 Å resolution (1). Its puzzlingly complex structure lacked the expected symmetry and regularity and launched the protein-folding problem. [With permission from the Medical Research Council Laboratory of Molecular Biology]
图 1 1958年, Kendrew及其合作者发表了球状蛋白的第一个结构: 6 Å分辨率下的肌红蛋白(1). 它的结构莫名其妙地复杂, 并缺乏预想的对称性和规则性. 这一事实开启了蛋白质折叠问题的研究.
"Perhaps the most remarkable features of the molecule are its complexity and its lack of symmetry. The arrangement seems to be almost totally lacking in the kind of regularities which one instinctively anticipates, and it is more complicated than has been predicated by any theory of protein structure. Though the detailed principles of construction do not yet emerge, we may hope that they will do so at a later stage of the analysis."
或许这种分子最引人注目的特征就是复杂性和缺乏对称性. 这种排列方式似乎完全缺少人们本能会期望的那种规则性, 它比任何一种蛋白质结构理论所预测的都要复杂. 尽管有关这个结构的详细构造原理还未发现, 我们仍希望在分析的后期阶段会进行这样的工作.The protein-folding problem came to be three main questions: (i) The physical folding code: How is the 3D native structure of a protein determined by the physicochemical properties that are encoded in its 1D amino-acid sequence? (ii) The folding mechanism: A polypeptide chain has an almost unfathomable number of possible conformations. How can proteins fold so fast? (iii) Predicting protein structures using computers: Can we devise a computer algorithm to predict a protein's native structure from its amino acid sequence? Such an algorithm might circumvent the time-consuming process of experimental protein-structure determination and accelerate the discovery of protein structures and new drugs. 蛋白质折叠问题主要包含三个问题: (i) 折叠的物理规则: 蛋白质一维氨基酸序列的理化性质如何决定蛋白质的3D天然结构? (ii) 折叠机制: 一个多肽链的可能构象不计其数. 蛋白质如何能折叠得这么迅速? (iii) 使用计算机预测蛋白质结构: 我们能不能设计一种计算机算法, 根据蛋白质的氨基酸序列来预测蛋白质的结构? 这种算法可避免实验测定蛋白质结构的费时过程, 从而加快蛋白质结构和新药物的发现. Here, we give our perspective on these questions at the broad-brush level. More detailed reviews can be found elsewhere (4-8). 本文中, 我们粗略地给出我们对这些问题的一些观点. 更多详细综述请参考(4-8).
The Physical Code of Protein Folding
蛋白质折叠的物理规则
What forces drive a protein to its 3D folded structure? Much insight comes from the Protein Data Bank (PDB), a collection of now more than 80,000 protein structures at atomic detail (9). The following factors appear to contribute (10): (i) Hydrogen bonds. Protein structures are composed of α-helices and β-sheets, as was predicted by Linus Pauling on the basis of expected hydrogen bonding patterns (11). (ii) van der Waals interactions. The atoms within a folded protein are tightly packed, implying the importance of the same types of close-ranged interactions that govern the structures of liquids and solids. (iii) Backbone angle preferences. Like other types of polymers, protein molecules have preferred angles of neighboring backbone bond orientations. (iv) Electrostatic interactions. Some amino acids attract or repel because of negative and positive charges. (v) Hydrophobic interactions. Proteins ball up into well-packed folded states in which the hydrophobic (H) amino acids are predominantly located in the protein's core and the polar (P) amino acids are more commonly on the folded protein's surface. Theory and experiments indicate that folding is governed by a predominantly binary code based on interactions with surrounding water molecules: There are few ways a given protein sequence of H and P residues can configure to bury its hydrophobic amino acids optimally (12, 13). (vi) Chain entropy. Opposing the folding process is a large loss in chain entropy as the protein collapses into its compact native state from its many open denatured configurations (12). 什么作用力驱使一个蛋白质形成其3D折叠结构? 许多洞察来自于蛋白数据库PDB. 目前其中已收集了80,000多个原子分辨率的蛋白质结构(9). 看来以下因素对蛋白质折叠有所贡献(10): (1) 氢键. 正如Linus Pauling(莱纳斯·鲍林)基于预期的氢键模式所预测的一样, 蛋白质结构由α螺旋和β折叠组成(11). (ii) 范德华相互作用. 折叠蛋白质中的原子紧紧地堆积在一起, 这暗示了相同类型的近程相互作用的重要性, 正是这种相互作用决定了液体和固体结构. (iii) 骨架的角度倾向性. 像其他类型的聚合物一样, 蛋白质分子相邻骨架键的取向也具有角度倾向性. (iv) 静电相互作用. 一些氨基酸由于带正电荷和负电荷而相互吸引或排斥. (v) 疏水作用. 蛋白质团成堆积很好的折叠状态, 其中疏水氨基酸(H, hydrophobic)主要位于蛋白质核心, 极性氨基酸(P, polar)通常更容易位于折叠蛋白质的表面. 理论和实验表明折叠主要是由二元规则控制, 基于与周围水分子的相互作用: 对于一个给定H和P残基的氨基酸序列, 只有很少几种方式能将其疏水氨基酸最优地隐埋在折叠结构中(12, 13). (vi) 链熵. 逆转折叠过程会损失大量的链熵, 因为蛋白质是从多种开放的变性构象转换为紧凑的天然状态的(12). These physical forces are described approximately by "forcefields" (14). Forcefields are models of potential energies that are used in computer simulations. They are widely applied to studies of protein equilibria and dynamics. In computer modeling, a protein molecule is put into an initial configuration, often random. Conformations change over the course of the simulation by repeatedly solving Newton's dynamical laws of motion for the atoms of the protein molecule and the solvent by using the forcefield energies. According to the laws of thermodynamics, systems tend toward their states of lowest free energy. Computational protein folding explores the process by which the protein proceeds through conformational states to states of lower free energies. As shown in Fig. 2, the thermodynamically stable states of 12 small protein structures can be reached fairly successfully by means of extensive molecular dynamics (MD) simulations in a bath of explicit water molecules (15). However, such successes, important as they are, are limited. So far, such modeling succeeds only on a limited set of small simple protein folds (16). And, it does not yet accurately predict protein stabilities or thermodynamic properties. Opportunities for the future include better forcefields, better models of the protein-water interactions, and faster ways to sample conformations, which are far too limited, even with today's most powerful computers. 这些物理作用力可以近似地由"力场"(forcefields)来描述(14). 力场是计算机模拟中使用的蛋白质的势能模型, 被广泛用于研究蛋白质的平衡以及动力学. 在计算机建模中, 蛋白质分子常常是随机地设为一个初始构象. 然后利用力场能量重复求解蛋白质分子和溶剂分子中原子的牛顿运动方程, 构象在模拟中就会发生改变. 根据热力学定律, 系统往往会趋向于其自由能最低的状态. 因此计算蛋白质折叠就是探索蛋白质从构象状态到最低自由能状态的过程. 如图 2 所示, 通过处于显式水分子环境中的大量分子动力学(MD, molecular dynamics)模拟, 可以相当成功地获得12个小型蛋白质的结构(15). 然而, 这些成功尽管重要却也很有限. 到目前为止, 这样的建模只在有限的小型简单蛋白质折叠过程中成功过(16). 并且, 它也还没有精确地预测出蛋白质的稳定性或热力学性质. 未来的发展方向包括更好的力场, 处理蛋白质与水相互作用的更好模型, 以及更快的构象采样方法. 即便是使用现今最高性能的计算机, 构象采样也受到很大的限制. The early days saw hopes of finding simple sequence patterns-say of hydrophobic, polar, charged, and aromatic amino acids--that would predict protein structures and stabilities. That has not materialized. Nevertheless, the results of the detailed atomic simulations described above give optimism that atomically detailed modeling is systematically improving and is contributing to our understanding of protein sequence-structure relationships. 早期, 我们曾希望发现简单的序列模式, 如疏水, 极性, 带电和芳香氨基酸, 并以此来预测蛋白质的结构及其稳定性. 这个希望并没有成真. 然而, 上述详细的原子模拟结果仍让我们觉得乐观, 原子分辨率的建模正在系统性地改进, 并不断加深了我们对蛋白质序列与结构关系的理解.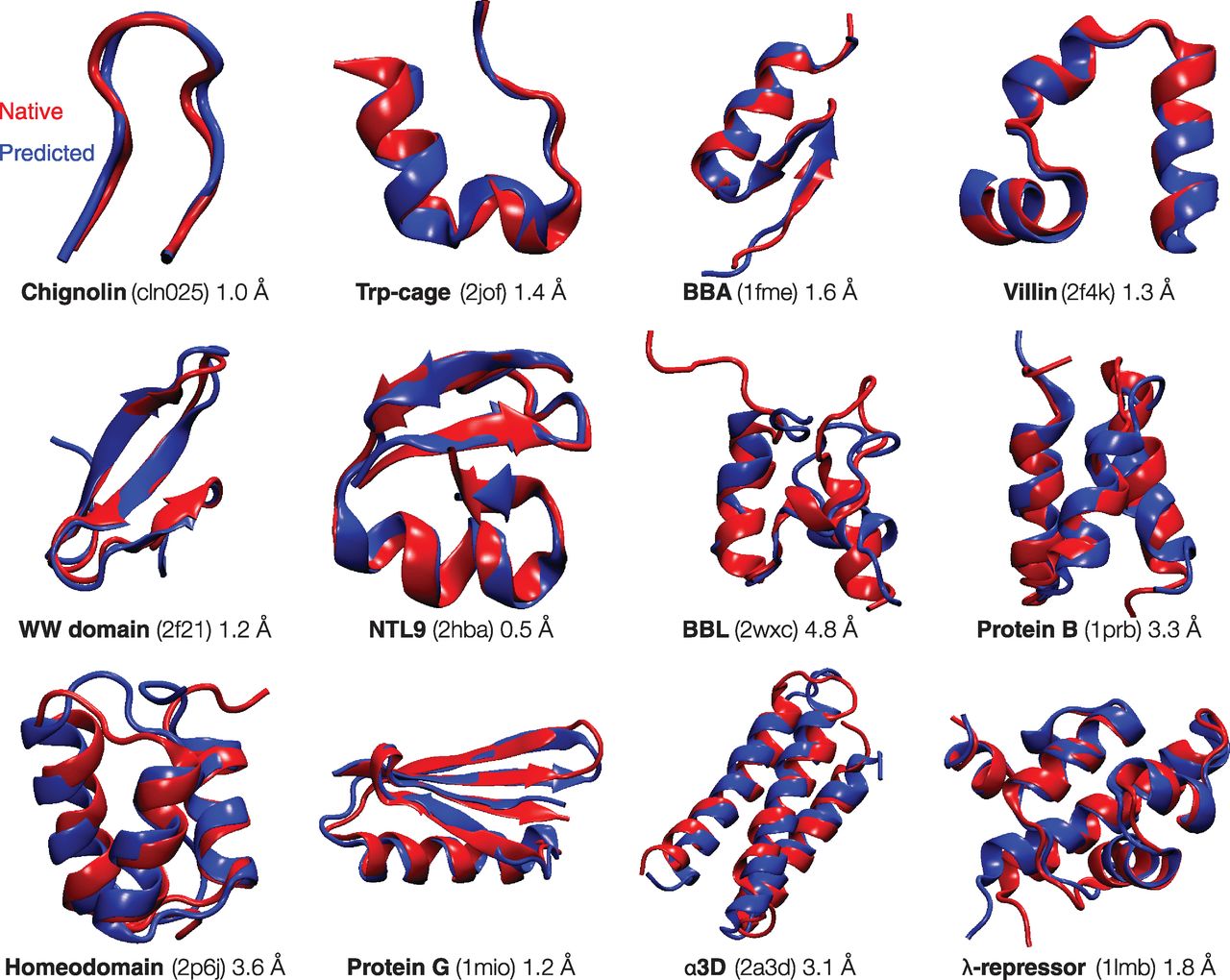 Fig. 2 Modern physical models can compute the folded structures of some small proteins. Using a high-performance custom computer called Anton (48), Shaw and co-workers observed reversible folding and unfolding in more than 400 events across 12 small proteins to structures within 4.5 Å of the experimental structure (15). The experimental structures are shown in red, and the computed structures are blue. Shown are the name, PDB identifier, and RMSD (root-mean-square deviation between alpha carbon atoms) between the predicted and experimental structures. [Adapted with permission (15)]
图 2 现代物理模型能够计算一些小型蛋白质的折叠结构. 使用定制的高性能Anton计算机(48), Shaw及其合作者对12个小型蛋白质观察到了400多次可逆的折叠和去折叠过程, 这些结构与实验结构的误差在4.5 Å以内(15). 实验结构如红色所示, 计算结构以蓝色表示. 图中显示了结构的名称, PDB标识符, 预测结构和实验结构之间的RMSD(α碳原子之间的均方根偏差).
Fig. 2 Modern physical models can compute the folded structures of some small proteins. Using a high-performance custom computer called Anton (48), Shaw and co-workers observed reversible folding and unfolding in more than 400 events across 12 small proteins to structures within 4.5 Å of the experimental structure (15). The experimental structures are shown in red, and the computed structures are blue. Shown are the name, PDB identifier, and RMSD (root-mean-square deviation between alpha carbon atoms) between the predicted and experimental structures. [Adapted with permission (15)]
图 2 现代物理模型能够计算一些小型蛋白质的折叠结构. 使用定制的高性能Anton计算机(48), Shaw及其合作者对12个小型蛋白质观察到了400多次可逆的折叠和去折叠过程, 这些结构与实验结构的误差在4.5 Å以内(15). 实验结构如红色所示, 计算结构以蓝色表示. 图中显示了结构的名称, PDB标识符, 预测结构和实验结构之间的RMSD(α碳原子之间的均方根偏差).
The Rate Mechanism of Protein Folding
蛋白质折叠的速率机制
At a meeting in Italy in 1968, Cyrus Levinthal raised the question (17) of how, despite the huge number of conformations accessible to it, a protein molecule can fold to its one precisely defined native structure so quickly (microseconds, for some proteins). How does the protein "know" what conformations not to search? 1968年在意大利举行的一个会议上, Cyrus Levinthal提出了这样一个问题(17), 尽管一个蛋白质拥有巨量的潜在构象, 但蛋白质如何能快速地折叠成精确定义的天然结构(对于一些蛋白质, 只需要几微秒). 蛋白质怎么不用通过搜索就"知道"那些构象的? This question led to a major experimental quest to characterize the kinetics of protein folding and to find folding intermediates, which are partially structured states along the "folding pathway" (18, 19). The hope was that snapshots of the chain caught in the act of folding would give insights into folding "mechanisms," the rules by which nature performs conformational searching. The experimental challenge was not just to measure atom-by-atom contacts within the heterogeneous interior of a protein molecule, but to do it on the fly, over microsecond-to-second time scales. This drove development of a powerful arsenal of new experimental methods, including mutational studies, hydrogen exchange, fluorescence labeling, laser temperature jumps, and single-molecule methods [reviewed elsewhere (7)]. 这个问题引发了一些主要的实验探究, 表征蛋白质折叠动力学的特征, 寻找折叠的中间体, 中间体是"折叠途径"上部分结构化的状态(18, 19). 人们希望能够捕捉到链在折叠过程中的快照, 并由此得以洞察折叠"机制", 也就是大自然进行构象搜索的规则. 这个实验的挑战不仅仅在于需要测量蛋白质分子异质内部所有原子间的接触, 还在于需要在微秒到秒的时间尺度内实时进行测量. 这推动了更强大的新实验方法的发展, 包括突变研究, 氢氘交换, 荧光标记, 激光温度跃变和单分子方法[参考综述(7)]. The general-principles solution of the needle-in-a-haystack conundrum emerged from polymer statistical thermodynamics. Studies of the chain entropies in models of foldable polymers showed that more compact, low-energy conformational ensembles have fewer conformations (12, 20-23), indicating that protein-folding energy landscapes are funnel-shaped (Fig. 3). Protein folding landscapes are narrower at the bottom; there are few low-energy, native-like conformations and many more open unfolded structures. A protein folds by taking random steps that are mostly incrementally downhill in energy. Steps need only be favorable by one to two times the thermal energy to reach the native structure rapidly (24). Insights from funnels, however, have not yet been sufficient to improve computer search methods. A landscape that appears smooth and funnel-shaped on a global scale can be rough on the local scales that are sampled in computer simulations. 在聚合物统计热力学中, 人们找到了一般原则性的解决方法, 意义解决这个大海捞针般的难题. 对可折叠聚合物模型中链熵的研究表明, 更紧密, 能量更低的构象系综包含的构象数更少(12, 20-23), 这表明蛋白质折叠的能量形貌呈漏斗状(图 3). 蛋白质折叠的形貌在底部更狭窄些; 低能量, 类天然结构的构象很少, 开放的, 未折叠的结构很多. 蛋白质通过随机步骤进行折叠, 并且大部分是沿着能量逐步降低的方向. 当能量达到热能的一到二倍时, 折叠步骤即可进行, 蛋白质能够迅速到达天然状态(24). 然而, 从漏斗形貌中得出的见解并不足以改进计算机使用的搜索方法. 从全局角度看, 形貌是光滑的, 呈漏斗状, 而从计算机模拟中进行采样的局域尺度来看, 形貌是很粗糙的. But we are still missing a "folding mechanism." By mechanism, we mean a narrative that explains how the time evolution of a protein's folding to its native state derives from its amino acid sequence and solution conditions. A mechanism is more than just the sequences of events followed by any one given protein in experiments or in computed trajectories. We do not yet have in hand a general principle that is applicable to a broad range of proteins, that would explain differences and similarities of the folding routes and rates of different proteins in advance of the data, and that properly average, in some meaningful way, over "irrelevant" thermal motions. One difficulty has been reconciling our "macroscopic" understanding of kinetics (mass-action models) that result from ensemble-averaged experiments with our "microscopic" understanding of the angstrom-by-angstrom changes of each protein conformation in computer simulations (energy landscapes). However, there are a few general conclusions (25). Proteins appear to fold in units of secondary structures. A protein's stability increases with its growing partial structure as it folds. And, a protein appears to first develop local structures in the chain (such as helices and turns) followed by growth into more global structures. Even though the folding process is blind, nevertheless it can be fast because native states can be reached by this divide-and-conquer, local-to-global process (26, 27). Funneled landscapes predict that the different individual molecules of the same protein sequence may each follow microscopically different routes to the same native structure. Some paths will be more populated than others. 但我们仍然缺少一种"折叠机制". 所谓机制, 我们指的是这样一个陈述, 解释了如何根据蛋白质的氨基酸序列及其所处的溶液环境, 推导出蛋白质折叠到天然状态的时间演化过程. 对于实验或计算轨迹中给定的一个蛋白质, 机制不仅仅是它所遵从的一系列事件. 现在我们还没有一个能够适用于大部分蛋白质的普遍原理. 在获得数据之前, 它就能够解释不同蛋白质折叠途径和折叠速率的差异与相似性. 它能够通过一些有意义的方式适当地平均化"不相关"的热运动. 困难之一是协调我们对动力学的"宏观"理解(质量作用模型)与对蛋白质每一构象一埃一埃变化的"微观"理解(能量形貌), 前者来自系综平均的实验, 后者来自计算机模拟. 然而, 我们有几个一般性的结论(25). 蛋白质似乎以二级结构为单位进行折叠. 蛋白质的稳定性随着它折叠部分的增加而增加. 并且, 蛋白质链中的局域结构(例如螺旋和转角)似乎首先发生变化, 然后更全局的结构才开始变化. 尽管折叠进程很盲目, 然而蛋白质却可以通过分而治之和局域到全局的过程快速形成其天然状态(26, 27). 漏斗状的形貌预测, 同一蛋白质序列的各个分子可能沿微观上不同的途径形成相同的天然结构. 一些途径上的分子要比其他途径上的多.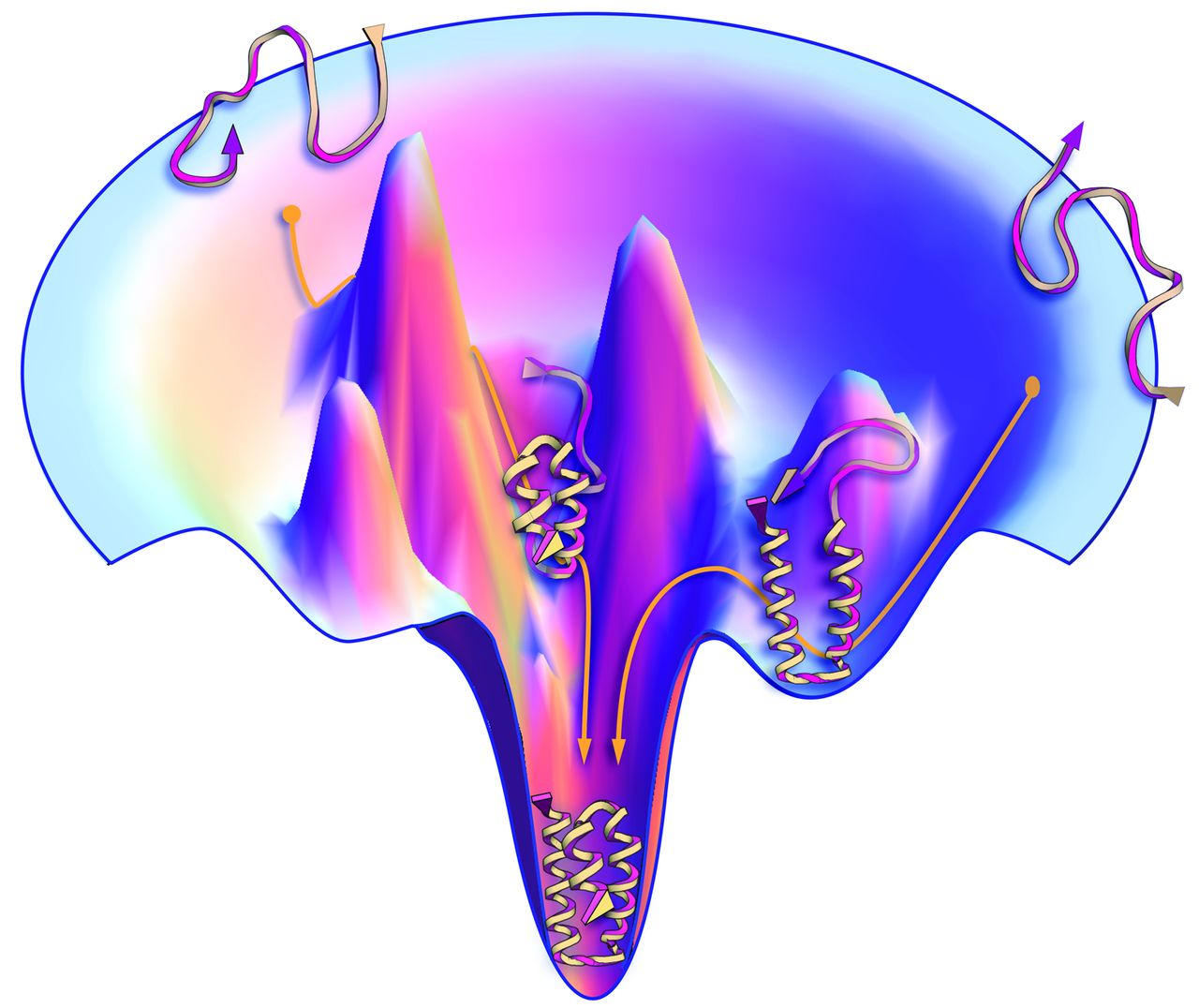 Fig. 3 Proteins have a funnel-shaped energy landscape with many high-energy, unfolded structures and only a few low-energy, folded structures. Folding occurs via alternative microscopic trajectories.
图 3 蛋白质有一个漏斗状的能量形貌, 其中有很多高能量的未折叠结构, 以及少量低能量的折叠结构. 折叠是通过各种微观轨迹发生的.
Fig. 3 Proteins have a funnel-shaped energy landscape with many high-energy, unfolded structures and only a few low-energy, folded structures. Folding occurs via alternative microscopic trajectories.
图 3 蛋白质有一个漏斗状的能量形貌, 其中有很多高能量的未折叠结构, 以及少量低能量的折叠结构. 折叠是通过各种微观轨迹发生的.
Computing Protein Structures from Amino Acid Sequences
根据氨基酸序列计算蛋白质的结构
A grand challenge has been to develop a computer algorithm that can predict a protein's 3D native structure from its amino acid sequence. On the one hand, knowledge of native structures is a starting point for understanding biological mechanisms and for discovering drugs that can inhibit or activate those proteins. On the other hand, we know 1000-fold more sequences than structures, and this gap is growing because of developments in high-throughput sequencing. So, there is considerable value in methods that could accurately predict structures from sequences. 开发一种根据氨基酸序列就能预测蛋白质的3D天然结构的计算机算法是一个巨大的挑战. 一方面, 天然结构的相关知识是理解生物机制, 发现抑制或激活蛋白质的药物的一个出发点. 另一方面, 我们所知道的序列是结构的1000倍, 而且由于高通量测序技术的发展, 这个差距还在增大. 因此, 根据序列准确预测结构的方法非常有价值. Computer-based protein-structure prediction has been advanced by Moult and colleagues, in an event initiated in 1994 called CASP: Critical Assessment of protein Structure Prediction (28, 29). Held every second summer, CASP is a community-wide blind competition in which typically more than 100 different "target sequences" (of proteins whose structures are known but not yet publicly available) are made available to a community that numbers more than 150 research groups around the world. Each participating group applies some algorithmic scheme that aims to predict the 3D structures of these target proteins. After each CASP event, the true experimental structures are then revealed, group performances are evaluated, and community evaluations are published. 通过在1994年发起一个叫CASP(蛋白质结构预测关键评估)的竞赛, Moult及其同事推进了基于计算机的蛋白质结构预测(28, 29). 每两年一次, 在夏季举行的CASP是一个全社会的无目的竞赛, 来自全世界的150多个研究组通常会得到100多个不同的"目标序列"(相应蛋白质的结构已知但尚未公布). 每个参赛小组运用一些算法方案来预测这些目标蛋白质的3D结构. 每次CASP结束后, 会公布这些蛋白质真正的实验结构, 并评估所有小组的表现, 出版评估结果. Currently, all successful structure-prediction algorithms are based on assuming that similar sequences lead to similar structures. These methods draw heavily on the PDB, which now contains more than 80,000 structures. However, many of these structures are similar, and the PDB contains only ~4000 structural families and 1200 folds (30). 当前, 所有成功的结构预测算法都基于相似序列有相似结构这个假设. 这些方法非常依赖于现已包含80,000多个结构的PDB. 然而, 这些结构中有很多是相似的, 因而PDB大约只包含4000个结构族和1200个折叠子(30). CASP-wide progress over the past 18 years is summarized in Fig. 4A. Prediction accuracies improved from CASP1 (1994) to CASP5 (2002) on the basis of several advances: (i) PDB expanded from ~1600 structures to 19,000 during that time. (ii) Better sequence search and alignment tools, such as Position-Specific Iterated Basic Local Alignment Search Tool (PSI-BLAST) (31), enabled the detection of more remote evolutionary relationships and more accurate sequence alignments. (iii) A strategy, called the "fragment assembly approach" (32-35), was developed that can often improve predictions when a similar sequence cannot be found in the PDB. CASP过去18年中所取得的进展总结在图 4A中. 从CASP1(1994年)到CASP5(2002年), 预测精度的提高是由于以下几方面的进步: (i) 在那段时间, PDB从约1600个结构扩大到19,000个. (ii) 更好的序列搜索和比对工具, 如置限定迭代的基本局部比对搜索工具(PSI-BLAST, Position-Specific Iterated Basic Local Alignment Search Tool)(31), 使得探测蛋白质更远的进化关系, 更准确的序列比对成为可能. (iii) "片段拼接"方法(32-35)的发展. 当在PDB中找不到相似序列时, 利用这个方法通常可以提高预测的精确性. If the target protein's sequence is related to a sequence that is already in the PDB, predicting its structure is usually easy (Fig. 4). In such cases, target protein structures are predicted by using "template-based modeling" (also called homology modeling or comparative modeling). But when there is no protein in the PDB with a sequence resembling the target's, accurately predicting the structure of the target is much more difficult. These latter predictions are called "free modeling" (also called ab initio or de novo prediction). One of the most successful free-modeling techniques is fragment assembly, described below. Many prediction methods are hybrids, combining template-based modeling, fragment assembly, and other strategies. 如果目标蛋白质序列与PDB中的已有序列有关, 那么预测它的结构通常很容易(图 4). 在这种情况下, 通常使用"基于模板的建模"方法(也称同源模建或比较建模)来预测目标蛋白质的结构. 但是当PDB中没有与目标序列相似的蛋白质序列时, 准确预测目标蛋白质的结构困难得多. 后一种预测方法叫做"自由建模"(也叫做从头预测). 最成功的自由建模技术之一是片段拼接, 将在下面进行说明. 许多预测方法都是混合算法, 结合了基于模板的建模方法, 片段拼接以及其他一些策略. In fragment assembly (32-35), a target protein sequence is deconstructed into small, overlapping fragments. A search of the PDB is performed to identify known structures of similar fragment sequences, which are then assembled into a full-length prediction. The qualities of fragments and their assemblies are assessed by using some form of scoring function that aims to select more native-like protein structures from among the many possible combinations. Problems of folding physics described above share more commonality with free modeling than with template-based modeling. 在片段拼接方法中(32-35), 目标蛋白质序列先被分解为一些小的, 相互重叠的片段. 然后在PDB进行搜索, 找出相似片段序列的已知结构, 最后拼接成一个全长的预测结构. 片段及其拼接物的质量用某种形式的打分函数来评估, 使用它的目的在于从许多可能的组合中挑选出更接近蛋白质天然结构的片段. 与基于模板的建模相比, 这种方法和自由建模具有更多的共同点. Since CASP6, although overall progress has slowed (Fig. 4A), there has been systematic, incremental progress (36). The best groups can now on average produce models that are better than the single best template from the PDB. Progress has been made toward successfully combining multiple templates into a single prediction. Substantial improvements have been observed for free-modeling targets shorter than 100 amino acids, although no single group yet consistently produces accurate models. Larger free-modeling targets remain challenging. Several recent algorithmic developments--to predict residue-residue contacts from sequence alone (37-39) and to more sensitively and accurately identify remote homologs (40)--promise to further improve prediction accuracy. 自CASP6以来, 尽管总体进展放缓(图 4A), 但仍存在系统性的, 循序渐进的进步(36). 目前, 表现最好的小组平均来说可以构建出比PDB中单个最好的模板还要好的模型. 现在已经成功地将多个模板组合应用到单个预测中. 尽管还没有单个小组能自洽地构建出精确的模型, 对短于100个氨基酸的目标蛋白质, 自由建模已经有了实际性的改善. 利用自由建模预测更大型的目标仍富有挑战性. 最近发展的几种算法--仅根据序列预测残基与残基的接触(37-39), 更敏感而准确地识别远亲同源蛋白(40)--有望进一步提高预测的精度. The performance of two of the best fully automated server predictors during CASP9 (41) are shown in Fig. 4B: HHPred, a pure template-based modeling tool (42), and ROSETTA, a hybrid tool that combines fragment assembly and template-based modeling with all-atom refinement (43). For some fraction of CASP targets [~10%, based on a cutoff of 85 Global Distance Test-Total Score (GDT-TS) (44), which is defined in Fig. 4, legend], the best predictions are now accurate enough to interpret biological mechanisms, to guide biochemical studies, or to initiate a drug-discovery program (which requires structural errors of less than 2 to 3 Å). However, it remains a challenge to predict the other 90% of protein structures this accurately. In addition, it is critical to also improve physics-based technologies and to reduce our dependence on knowledge of existing structures, so that we can ultimately study protein motions, intrinsically disordered proteins, induced-fit binding of drugs, and membrane proteins and foldable polymers, for which databases are too limited. 图 4B展示了CASP9期间两个最好的全自动预测服务器的表现: HHPred, 单纯基于模板的预测工具(42)和ROSETTA, 结合了片段拼接, 模板建模和全原子精修的混合预测工具(43). 对于CASP中的一部分目标蛋白质[约10%, 基于截断为85的全局距离完全测试得分(GDT-TS, Global Distance Test-Total Score)(44), 定义见图 4的图例], 目前的最好预测足够精确, 足以用于阐述生物学机制, 引导生物化学的研究, 或开启一个药品研发项目(要求结构误差小于2~3 Å). 然而, 以这个精度去预测其余90%的蛋白质的结构仍然是一个挑战. 另外, 增强基于物理的预测技术, 减少对已有结构的依赖性也很重要, 这样最终我们才能研究蛋白质的运动, 固有无序的蛋白, 药物的诱导结合, 以及膜蛋白和可折叠聚合物, 对这两种物质, 数据库非常有限.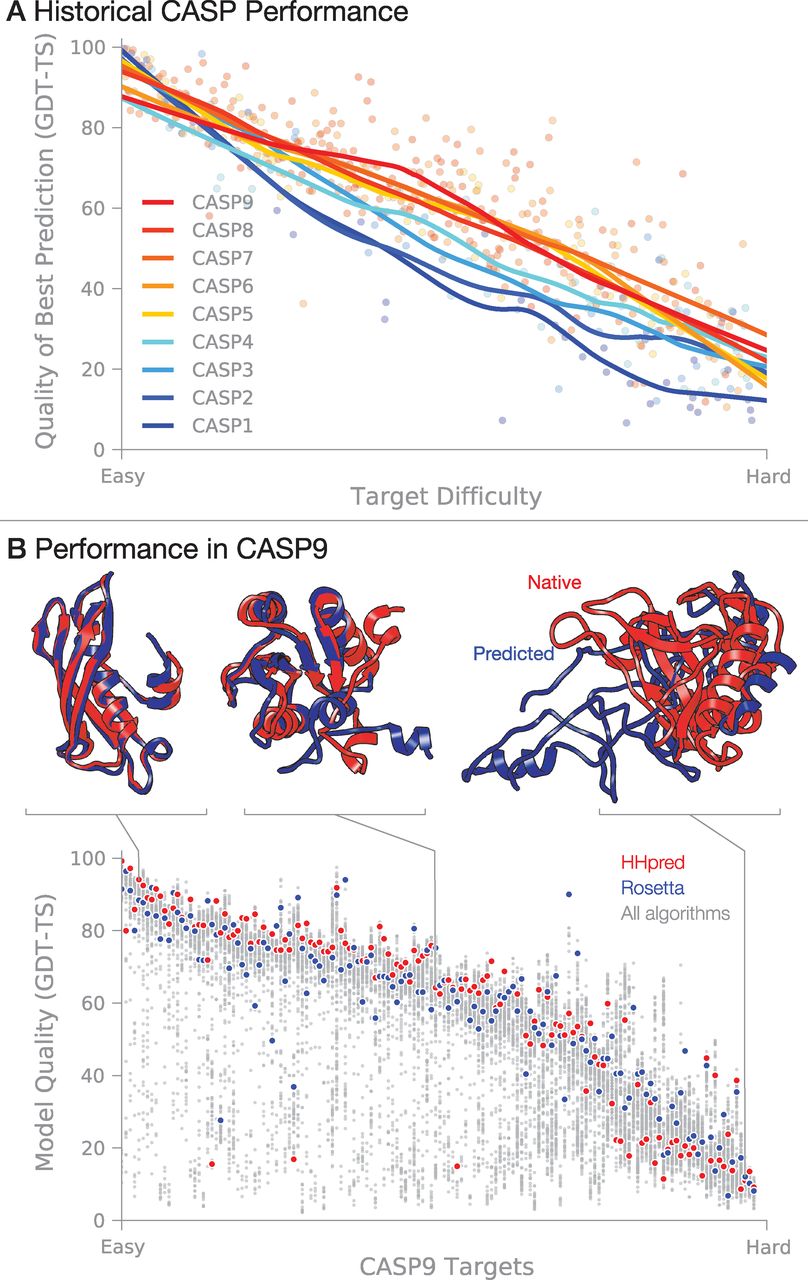 Fig. 4 Historical and present performance in CASP. Model quality is judged by using GDT-TS (44), which is approximately the percentage of residues that are located in the correct position. (A) Evolution of accuracy over the history of CASP, spanning 18 years. Each target is classified according to an approximate measure of difficulty that incorporates both the structural and sequence similarity to proteins of known structure (36). Each dot represents the best prediction (across all participants) for a given target. (B) Summary of prediction accuracy in CASP9 (41). We highlight the performance of two of the best automated server algorithms. Selected predictions are superimposed on the corresponding native structures to give a visual sense of the accuracy level that can be expected.
图 4 CASP的历史表现和当前表现. 模型的质量以GDT-TS来衡量(44), 它大致是指位于正确位置的残基的百分比. (A) CASP 18年来精度的变化. 每个目标大致是根据难度来分类的, 难度考虑了结构和序列与已知结构蛋白的相似性(36). 每个点代表一个给定目标的最佳预测(所有参与者中的). (B) CASP9预测精度的总结(41). 我们高亮了两个性能最好的全自动服务算法的性能. 所选的预测叠加在对应的天然结构上给出了所期望精度水平的直观感受.
Fig. 4 Historical and present performance in CASP. Model quality is judged by using GDT-TS (44), which is approximately the percentage of residues that are located in the correct position. (A) Evolution of accuracy over the history of CASP, spanning 18 years. Each target is classified according to an approximate measure of difficulty that incorporates both the structural and sequence similarity to proteins of known structure (36). Each dot represents the best prediction (across all participants) for a given target. (B) Summary of prediction accuracy in CASP9 (41). We highlight the performance of two of the best automated server algorithms. Selected predictions are superimposed on the corresponding native structures to give a visual sense of the accuracy level that can be expected.
图 4 CASP的历史表现和当前表现. 模型的质量以GDT-TS来衡量(44), 它大致是指位于正确位置的残基的百分比. (A) CASP 18年来精度的变化. 每个目标大致是根据难度来分类的, 难度考虑了结构和序列与已知结构蛋白的相似性(36). 每个点代表一个给定目标的最佳预测(所有参与者中的). (B) CASP9预测精度的总结(41). 我们高亮了两个性能最好的全自动服务算法的性能. 所选的预测叠加在对应的天然结构上给出了所期望精度水平的直观感受.
Protein Folding: The Legacy of a Basic Science
蛋白质折叠: 基础科学的遗产
Protein folding is a quintessential basic science. There has been no specific commercial target, yet the collateral payoffs have been broad and deep. Specific technical advances are reviewed elsewhere (7); below, we describe a few general outgrowths. 蛋白质折叠是典型的基础科学. 没有特定的商业目标, 然而其成果却广泛而深入. 具体的技术进展可参考(7). 下面, 我们描述几个一般性的成果. Growth of protein-structure databases. Today, more than 80,000 protein structures are known at atomic detail and publically available through the PDB. New structures are being added at a rapid pace, supported by the National Institutes of Health (NIH)-funded Protein Structure Initiative, which was developed in part to inform protein structure prediction. 蛋白质结构数据库的增长. 今天, 已经在原子细度上知道了超过80,000个蛋白质的结构, 并可通过PDB获得这些结构. 新结构正快速地添加到数据库中, 这是由美国国立卫生研究院(NIH, National Institutes of Health)资助的蛋白质结构项目支持, 在某种程度上, 这也是发展蛋白质结构的预测. Advances in computing technology. Understanding protein folding was a key motivation for IBM's development of the Blue Gene supercomputer (45), now also used to study the brain, materials, weather patterns, and quantum and nuclear physics. Protein folding has also driven key advances in distributed-grid computing, such as in Folding@home, developed by Pande at Stanford, in which computer users all over the world donate their idle computer time to perform physical simulations of protein systems (46). Folding@home, which now has more than one million registered users and an average of 200,000 user-donated CPUs available at any one time, provided some of the earliest simulations showing that MD simulations can accurately predict folding rates (47). The Anton computer from DE Shaw Research, custom designed to simulate biomolecules, gives several orders of magnitude better performance than conventional computers (48). Advances in computer technology have led to major advances in forcefields and to more reliable atomic-level insights into biological mechanisms. 计算技术的进步. 理解蛋白质折叠是IBM发展蓝色基因超级计算机(45)的主要动机, 现在它也被用于研究大脑, 材料, 气候模式, 以及量子物理学和核物理. 蛋白质 折叠也推动了分布式计算的重要进步, 比如通过由斯坦福大学Pande创立的Folding@home, 世界各地的计算机用户可以贡献自己电脑的闲置时间来进行蛋白质系统的物理模拟(46). Folding@home现在已有超过一百万的注册用户, 在任何时间平均有200,000个用户捐赠的CPU可供使用. Folding@home提供了一些最早的模拟, 结果表明分子动力学模拟可以准确地预测折叠速率(47). DE Shaw研究所的Anton计算机是专门设计用于生物分子模拟的, 它的性能比传统的计算机好出几个数量级(48). 计算机技术的进步带来了力场的重大进步以及在更可靠的原子层面对生物学机制的理解. Improvements in biomolecular forcefields. Computer processing power has advanced at the Moore's law rate, doubling every ~2 years. But equally important, forcefields have kept pace. Increased computer power leads to longer computed time scales, which puts more stringent demands on the accuracies of biomolecular forcefields. In a pioneering paper in 1977, McCammon et al. showed that the BPTI protein was stable in computer simulations during a computed time of 10 ps (49). Today, small proteins are typically stable in explicit-water simulations for 5 to 8 orders of magnitude longer--microseconds to milliseconds of computed time (50). Achieving such advances has required continuous improvements in forcefield accuracy. 生物分子力场的改进. 计算机处理能力的增长速率遵循摩尔定律, 大约每2年增加一倍. 但同样重要的是, 力场也保持了同样的增长速率. 计算机能力的增强使得计算的时间尺度更大, 也对生物分子力场的精度提出了更严格的要求. 在1977年的一篇开创性论文中, McCammon等人的研究表明, BPTI蛋白质在时长为10 ps的计算机模拟中是稳定的(49). 现今, 从微秒到毫秒, 在5到8个数量级的计算时间内, 小蛋白质在显式水模拟中一般是稳定的(50). 实现这种进步要求不断提升力场的精度. New sociological structures in the scientific enterprise. Protein folding has driven innovations in how science is done. CASP was among the first community-wide scientific competitions/collaborations, a paradigm for how grand-challenge science can be advanced through an organized communal effort. Other such competitions have followed, including Critical Assessment of Prediction of Interactions (CAPRI) (predicting protein-protein docking) (51), SAMPL (predicting small-molecule solvation free energies, and ligand binding modes and affinities) (52), and GPCR-Dock (predicting structures for G-protein coupled receptors, a pharmaceutically important category of membrane proteins) (53), among many others. Protein folding has also pioneered "citizen science," such as in Folding (46) and Robetta@home and in a computer game called Foldit (54), in which the public engages in protein folding on their home computers. 科技事业的新社会学结构. 蛋白质折叠推动着如何进行科学研究方面的创新. CASP是最早覆盖全社区的科学竞赛/协作之一, 通过有组织的集体努力对一个颇具挑战的科学取得进步的范例. 之后也有一些这样的竞赛, 其中包括相互作用预测关键评估(CAPRI, 预测蛋白质与蛋白质的对接)(51), SAMPL(预测小分子的溶剂化自由能, 以及配体 结合的模式和亲疏性)(52)和GPCR-Dock(预测G-蛋白耦合受体的结构, 这是制药学中的重要膜蛋白种类)(53). 蛋白质折叠也是"公众科学"的先驱, 如在Folding(46)和Robetta@home, 以及一款叫做Foldit的电脑游戏(54)中, 公众将他们的家用电脑用于研究蛋白质折叠. New materials: Sequence-specific foldable polymers. The principles and algorithms developed for protein folding have led to nonbiological, human-made proteins and to new types of polymeric materials. In particular, proteins have been designed that bind to and inhibit other proteins (fig. 5A) (55), have new folds (56), have new enzymatic activities (57), and act as potential new vaccines (58). Also, a class of nonbiological polymers has emerged, called "foldamers," that are intended to mimic protein structures and functions (59-62). Foldamers already have broad-ranging applications (63-67) as inhibitors of protein-protein interactions, broad-spectrum antibiotics, lung surfactant mimics, optical storage materials, a zinc-finger-like binder, an RNA-protein binding disrupter for application in muscular dystrophy, gene transfection agents, and "molecular paper" (Fig. 5B). Although such materials have potential applications in biomedicine and materials science, they also provide a way for us to test and deepen our understanding of protein folding. 新材料: 特定序列的可折叠聚合物. 为研究蛋白质折叠而发展的原理和算法启发人们制造了非生物的人造蛋白质和新型聚合物材料. 特别是, 人们设计了一些能够结合和抑制其他蛋白的蛋白质(如图 5A)(55), 有新型折叠的蛋白质(56), 有新的酶活性的蛋白质(57), 和能作为潜在的新疫苗的蛋白质(58). 并且, 还出现了一类非生物的聚合物, 称为"非天然折叠体", 它们旨在模仿蛋白质的结构和功能(59-62). 非天然折叠体已经有了广泛应用(63-67): 作为蛋白质与蛋白质相互作用的抑制剂, 广谱抗生素, 肺的表面活化剂仿造物, 光存储材料, 类锌指结合剂, 用于肌肉萎缩症的RNA蛋白的结合破坏剂, 转基因载体和"分子纸"(图 5B). 这些材料不仅在生物医学和材料科学有着潜在的应用价值, 也为我们提供了一 种测试和加深理解蛋白质折叠的途径. Protein-folding diseases. Protein-folding research began before it was known that there are diseases of protein folding. Before 1972, it was assumed that all infectious diseases were transmitted through the DNA and RNA carried by viruses and bacteria. But Prusiner's studies of a patient with Creutzfeldt-Jakob disease (CJD) led to a previously unrecognized disease mechanism--namely, protein misfolding (68). Protein misfolding is now known to be important in many diseases, including CJD and type II diabetes, as well as neurodegenerative diseases such as Alzheimer's, Parkinson's, Huntington's, and amyotrophic lateral sclerosis. The protein-folding enterprise has provided important underpinnings for our understanding of folding diseases. 蛋白质折叠疾病. 在知道蛋白质折叠会带来疾病之前, 对蛋白质折叠的研究就已经开始了. 1972年以前, 人们认为传染病都是通过病毒和细菌所携带的DNA和RNA进行传播的. 但Prusiner在研究一个克雅氏病(CJD, Creutzfeldt-Jakob disease)患者时发现了一种从未识别的疾病机制, 就是蛋白质错误折叠(68). 现在人们知道了蛋白质错误折叠在许多疾病中的重要性, 这些疾病包括克雅氏病和II型糖尿病, 以及一些神经退行性疾病, 如阿尔茨海默氏症, 帕金森氏症, 亨廷顿氏症和肌萎缩性脊髓侧索硬化症. 蛋白质折叠研究对我们理解折叠疾病提供了重要的基础.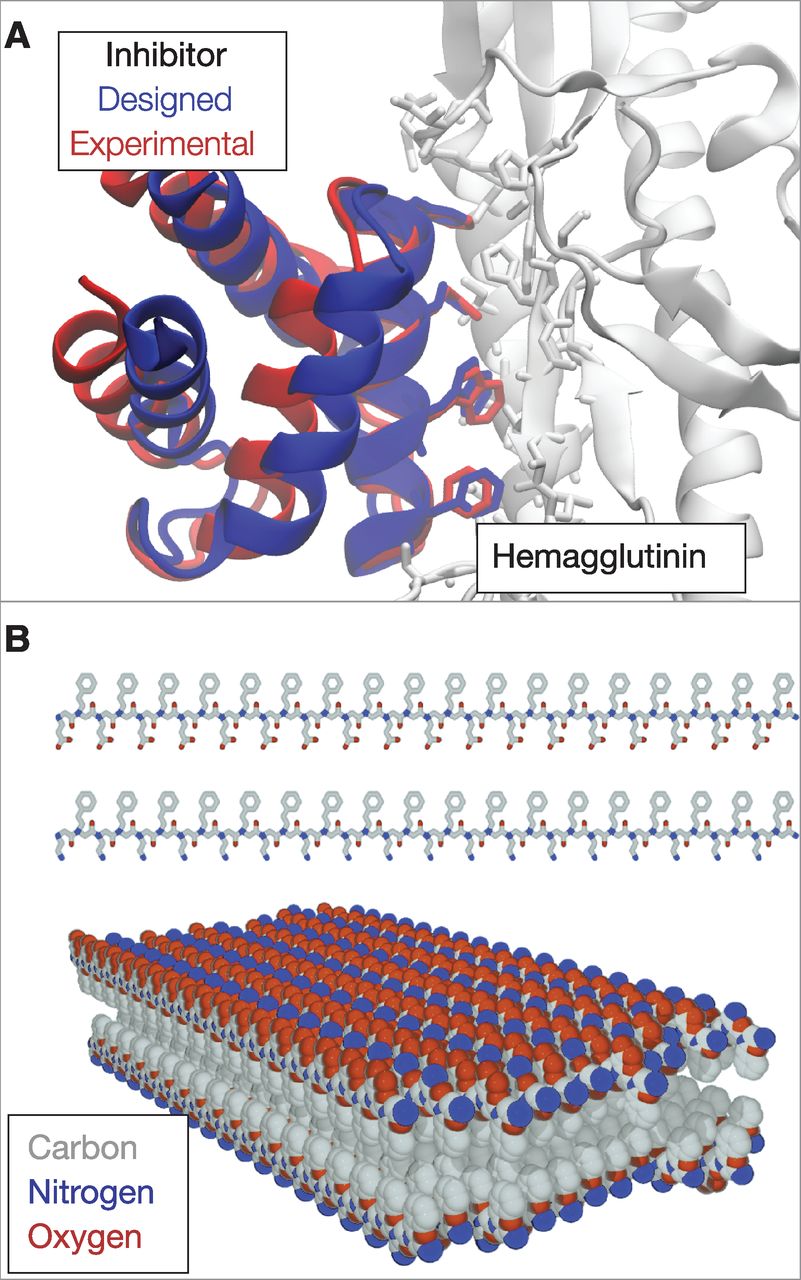 Fig. 5 Designed proteins and foldamers. (A) A protein inhibitor that was designed by computer to bind to hemagglutinin, an influenza protein. After design, the inhibitor was crystallized in a complex with hemagglutinin. The designed structure is in remarkably good agreement with experiment, particularly for the side chains involved in binding. [Adapted with permission (55)] (B) Peptoids are synthetic, foldable, protein-inspired polymers that have various applications. Shown here are peptoids that were designed as chains of alternating hydrophobic (gray) and either positively (blue) or negatively (red) charged side chains that spontaneously form a thin 2D structure called molecular paper. [Reprinted by permission (67)]
图 5 设计的蛋白质和非天然折叠体. (A) 由计算机设计, 用于结合一种流感蛋白--血凝素的蛋白质抑制剂. 设计后, 抑制剂在一个含有血凝素的复合物中进行结晶. 设计的结构与实验结构符合得非常好, 特别是对于涉及结合的侧链. (B) 拟肽是受蛋白质启发而合成的可折叠聚合物, 拥有各种用途. 这里展示的是设计为交替疏水(灰色)和带正电荷(蓝色)或负电荷(红色)侧链的拟肽, 它可自发形成一种叫做"分子纸"的二维薄层结构.
Fig. 5 Designed proteins and foldamers. (A) A protein inhibitor that was designed by computer to bind to hemagglutinin, an influenza protein. After design, the inhibitor was crystallized in a complex with hemagglutinin. The designed structure is in remarkably good agreement with experiment, particularly for the side chains involved in binding. [Adapted with permission (55)] (B) Peptoids are synthetic, foldable, protein-inspired polymers that have various applications. Shown here are peptoids that were designed as chains of alternating hydrophobic (gray) and either positively (blue) or negatively (red) charged side chains that spontaneously form a thin 2D structure called molecular paper. [Reprinted by permission (67)]
图 5 设计的蛋白质和非天然折叠体. (A) 由计算机设计, 用于结合一种流感蛋白--血凝素的蛋白质抑制剂. 设计后, 抑制剂在一个含有血凝素的复合物中进行结晶. 设计的结构与实验结构符合得非常好, 特别是对于涉及结合的侧链. (B) 拟肽是受蛋白质启发而合成的可折叠聚合物, 拥有各种用途. 这里展示的是设计为交替疏水(灰色)和带正电荷(蓝色)或负电荷(红色)侧链的拟肽, 它可自发形成一种叫做"分子纸"的二维薄层结构.
Unsolved Problems of Protein Physical Science
蛋白质物理学中的未解问题
Is the protein-folding problem "solved" yet (69)? We believe it is no longer useful to frame the question this way. Protein folding is now a whole field of research--a large, growing, and diverse enterprise. A field of science--such as physics, chemistry, or biology--is bigger than any individual research question. A field is self-perpetuating; a few old puzzles generate more new puzzles. For the field of protein physical science, the future is at least as compelling as the past. Here are some of the unsolved problems: 蛋白质折叠问题已经"解决"了吗(69)? 我们认为这样的问题不再有意义. 蛋白质折叠现在已经成为一个巨大的, 不断增长的, 多种多样的研究领域. 它是一个包含了物理, 化学或生物的科学领域, 比任何一个个别的问题都要宽泛. 这是一个自身能够维持其存在的领域, 一些旧问题能够带来更多的新问题. 对蛋白质物理学领域, 未来至少会和过去一样引人入胜. 下面是一些尚未解决的问题:- We have little experimental knowledge of protein-folding energy landscapes.
- We cannot consistently predict the structures of proteins to high accuracy.
- We do not have a quantitative microscopic understanding of the folding routes or transition states for arbitrary amino acid sequences.
- We cannot predict a protein's propensity to aggregate, which is important for aging and folding diseases.
- We do not have algorithms that accurately give the binding affinities of drugs and small molecules to proteins.
- We do not understand why a cellular proteome does not precipitate, because of the high density inside a cell.
- We know little about how folding diseases happen, or how to intervene.
- Despite their importance, we still know relatively little about the structure, function, and folding of membrane proteins (70, 71).
- We know little about the ensembles and functions of intrinsically disordered proteins (72), even though nearly half of all eukaryotic proteins contain large disordered regions. This is sometimes called the "protein nonfolding problem" or "unstructural biology."
- 关于蛋白质折叠的能量形貌, 实验上我们知之甚少.
- 我们不能一致地以高精度预测蛋白质的结构.
- 对任意氨基酸序列的折叠途径或过渡态, 我们还没有定量的微观理解.
- 我们不能预测蛋白质的聚集倾向, 而它对衰老和折叠疾病都非常重要.
- 我们还没有一个算法, 能够准确地给出药物或小分子与蛋白质的结合能力.
- 我们还不理解为什么在高密度的细胞内, 细胞的蛋白质组也不沉淀.
- 我们几乎不知道折叠疾病是如何发生的, 或者该如何进行干预.
- 尽管膜蛋白很重要, 我们对其结构, 功能和折叠的了解仍然相对较少(70, 71).
- 我们对内在无序蛋白质的整体和功能知之甚少(72), 尽管有近一半的真核蛋白质都含有大的无序区域. 这有时也被称为蛋白无折叠问题或无结构生物学.
Summary
总结
Fifty years ago, the protein-folding problem was born as a grand challenge of basic science. Since then, our understanding has advanced considerably. And, outgrowths of protein folding include the commercial development of new computers, such as IBM's Blue Gene; new modes of citizen science, including Folding@Home and Foldit; the development of communal scientific competitions, such as CASP; a database of now more than 80,000 protein structures; the Moore's-Law advancement in biomolecular simulation forcefields; new areas of materials science based on foldable polymers; and a foundation for understanding whole new classes of diseases--such as Alzheimer's, Parkinson's, and type II diabetes, called folding diseases--that were not even known when the protein-folding problem was first identified. 五十年前, 蛋白质折叠问题作为基础科学的巨大挑战而诞生. 从那时起, 我们对它的理解大大加深了. 由此引发的成果, 包括新型计算机的商业开发, 如IBM的蓝基因; 公众科学的新模式, 包括Folding@Home和Foldit; 公共科学竞赛的发展, 如CASP; 现已超过80,000个蛋白质结构的数据库; 生物分子模拟力场遵循摩尔定律的发展; 基于可折叠聚合物的材料科学的新领域; 以及理解新型疾病--如阿尔茨海默氏症, 帕金森氏症和II型糖尿病等所谓的蛋白质折叠疾病--的基础, 在蛋白质折叠问题被确认前我们甚至都不知道这些疾病. In times when there are pressures on science budgets for immediate payoffs, it is worth repeating the well-worn point that untargeted basic science often pays off in unexpected ways. 在科学预算顶着及时回报压力的今天, 有必要重复这个老生常谈的观点: 无针对性的基础科学常会以意想不到的方式回报投资.评论
- 2016-09-14 16:41:41
吴思晋李老师给力 -
2016-09-15 02:51:21
Jerkwin谢谢鼓励. - 2016-06-21 06:32:30
Will很不错,谢谢分享编译 -
2016-06-21 08:58:12
Jerkwin希望你也能有贡献. - 2016-07-08 20:08:00
赤道往北21°上次刚听了一个蛋白质折叠的讲座,里面还提到了这篇文章,正想看呢,感谢翻译菌 -
2016-07-09 03:26:32
Jerkwin不客气, 希望能对你有帮助. - 2017-04-04 11:14:15
周峰感谢🙏
