- 2016-09-23 20:15:59
在涉及固体和液体的计算中, 一个体系中通常会包含多个相同的分子. 这些分子加入时使用的方法不同, 其原子的排列次序可能很乱, 相互之间的对应次序也会不同. 而在做计算时, 我们习惯让这些分子按顺序排列, 属于同一个分子的原子依次排列, 而且不同分子间的原子一一对应, 这样在编程处理时会方便很多.
上面的论述可能有点抽象, 下面是一个例子.
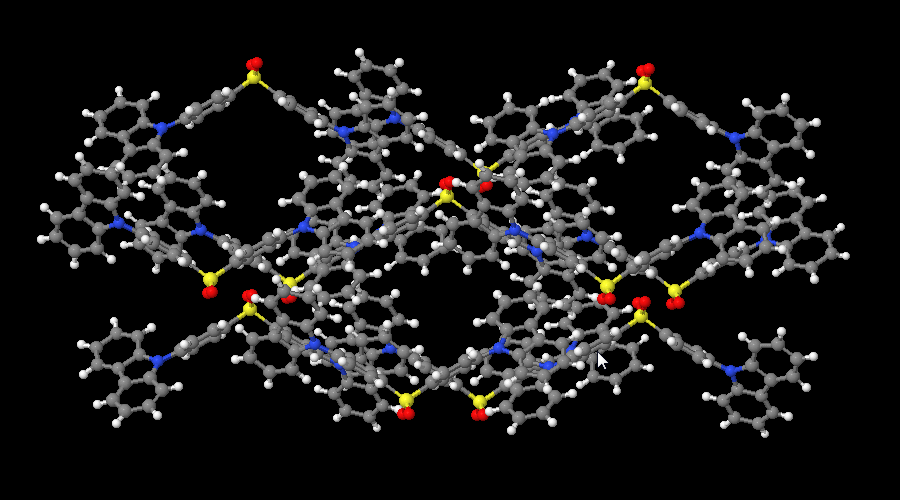
这是一个由晶体文件创建的体系, 里面包含12个分子, 每个分子又包含65原子.
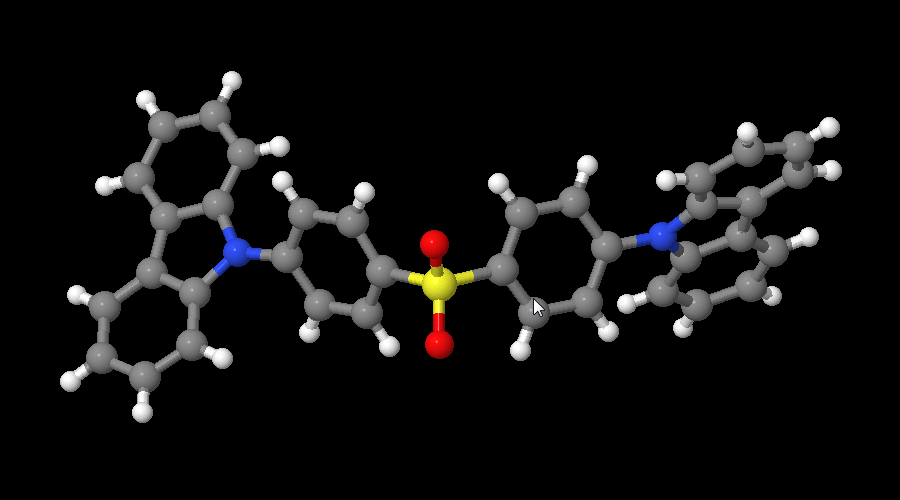
这些原子的排列顺序是乱的, 隶属于同一个分子的原子会分散到多处, 而不是集中在一起. 我们想要对这些原子的排列顺序进行整理, 使得首先是属于第一个分子的原子, 然后是属于第二个的, 第三个的…, 而且最好不同分子之间的原子可以一一对应好.
第一个目的相当于对整个体系进行片段分析, 根据原子间的连接性将原子划分到不同的分子中. 只要知道了原子间的连接性, 可以使用图论中的遍历算法来完成. 具体来说可采用广度优先的遍历方法(BFS). 这个算法还有其他的名字, 比如最大相连子图, 连接元件等, 是个经典算法. 详细的说明网上很多资料.
将原子划分为不同的分子后, 不同分子间的原子如何一一对应呢? 这个问题本质上属于广义的匹配问题. 二维图的匹配已经是NP难度的了, 再考虑三维结构的匹配就更加困难了. 虽然有一些算法可以解决, 但分子中的原子数多了以后就不适用了, 而且当分子间的差异过大时, 没有算法可以保证一定有效.
但当各个分子间的差异不大时, 我想到的方法有下面几种:
- 先根据连接性进行子图匹配, 列出所有可能的匹配方案, 然后再根据叠合后的RMSD来选择最佳匹配. 这种方法很严格, 但可能很耗时.
- 先对分子进行旋转, 使不同分子的取向一致, 然后根据距离最小规则进行原子匹配. 确定如何旋转时, 可使用某些特征量做为坐标轴, 如惯性主轴, 电四极矩主轴等, 也可手动选择不共线的三个特征原子, 以它们确定坐标轴. 前一方法对高对称性的分子可能会有问题, 而后一方法需要手动操作, 各有优缺点, 应根据实际情况选用.
- GaussView软件的
Edit | Connection提供了一个基于连接信息的匹配功能. 如果分子没有对称性的话, 基本可以正确匹配, 但当分子具有对称性时, 会存在多种匹配可能, 给出的匹配可能有误.
总言之, 上面的这些方法, 无论哪种都不能保证结果一定合理, 但至少可以提供一个比较合理的初步匹配, 供你最后手动调整一下.
Talk is cheap, show me the CODE
下面的代码用于对*.gjf格式的文件进行片段分析, 这种文件在最后可以保留原子间的链接信息. 当然, 稍加修改就可以用于其他格式的文件, 如pdb, mol2等. 代码最后会将所有的分子片段输出到一个*~Frg.xyz文件, 用于检查, 同时还会将每个分子片段分别输出到一个*~#.gjf文件, 用来提供给GaussView以便使用其Edit | Connection功能.
| FrgMol.bsh | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 | usage="\
>>>>>>>>>>>>>>>> FrgMol <<<<<<<<<<<<<<<<
>>>>>>>>>>>>>>>> Jicun LI <<<<<<<<<<<<<<<<
>>>>>>>>>> 2016-09-22 16:19:51 <<<<<<<<<
>> Usage: FrgMol <File.gjf>"
[[ $# -lt 1 ]] && { echo "$usage"; exit; }
File=${1%.gjf}
awk -v File=$File ' BEGIN{ Nblk=0; Ntot=0 }
NF==0 && Nblk<=2 { Nblk++; next }
Nblk==2 {
while(NF>0) {
getline; Ntot++
S[Ntot]=$1; X[Ntot]=$2; Y[Ntot]=$3; Z[Ntot]=$4
}
Ntot--
for(i=1; i<=Ntot; i++) {
getline
Imol[i]=0; Adj[i,i]=1
for(k=1; k<=(NF-1)/2; k++) { Adj[i,$(2*k)]=1; Adj[$(2*k),i]=1 }
}
Nmol=0
for(i=1; i<=Ntot; i++) {
if(Imol[i]==0) {
Nmol++; que[i]++
while(length(que)>0) {
for(v in que) {
Imol[v]=Nmol; Natm[Nmol]++
for(j=1; j<=Ntot; j++) {
if(Adj[v,j] && Imol[j]==0) que[j]++
}
delete que[v]
}
}
}
}
for(j=1; j<=Nmol; j++) {
print Natm[j]
print "Mol " j "/"Nmol
Fout=File"~"j".gjf"
print "# \n\nFrg "j"/"Nmol"\n\n0 1" >Fout
for(i=1; i<=Ntot; i++) {
if(Imol[i]==j) {
printf "%4s %14.8f %14.8f %14.8f\n", S[i], X[i], Y[i], Z[i]
printf "%4s %14.8f %14.8f %14.8f\n", S[i], X[i], Y[i], Z[i] > Fout
}
}
}
}
' $File.gjf > $File~Frg.xyz
|
如果需要测试的话, 点击这里下载脚本和测试文件吧.
GaussView连接匹配功能的使用
官网的说明见The Connection Editor, 下面以上面给出的测试文件示例一下.
运行bash FrgMol.bsh Mol.gjf会得到Mol~Frg.xyz和Mol~1.gjf, Mol~2.gjf, …Mol~12.gjf.
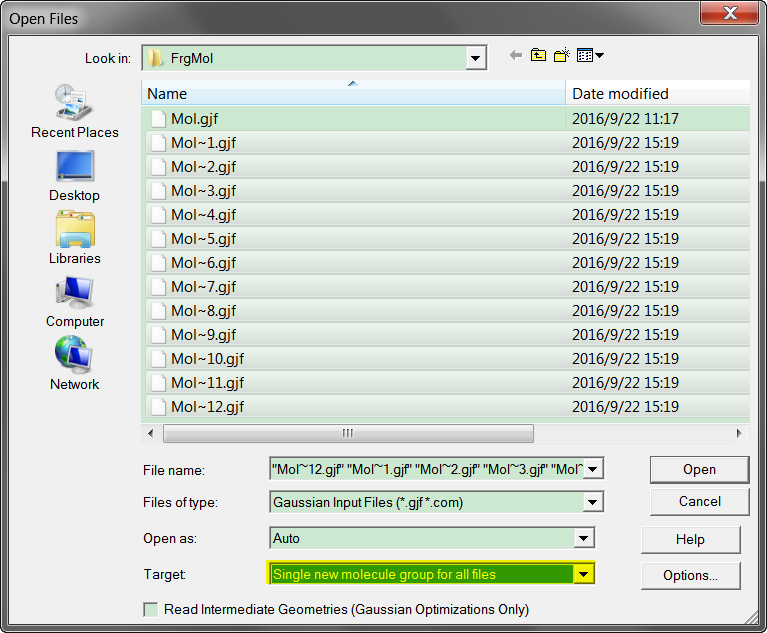
打开GaussView, 点击File | Open..., 按住Shift键, 选中12个gjf文件, 注意最下面的选择.
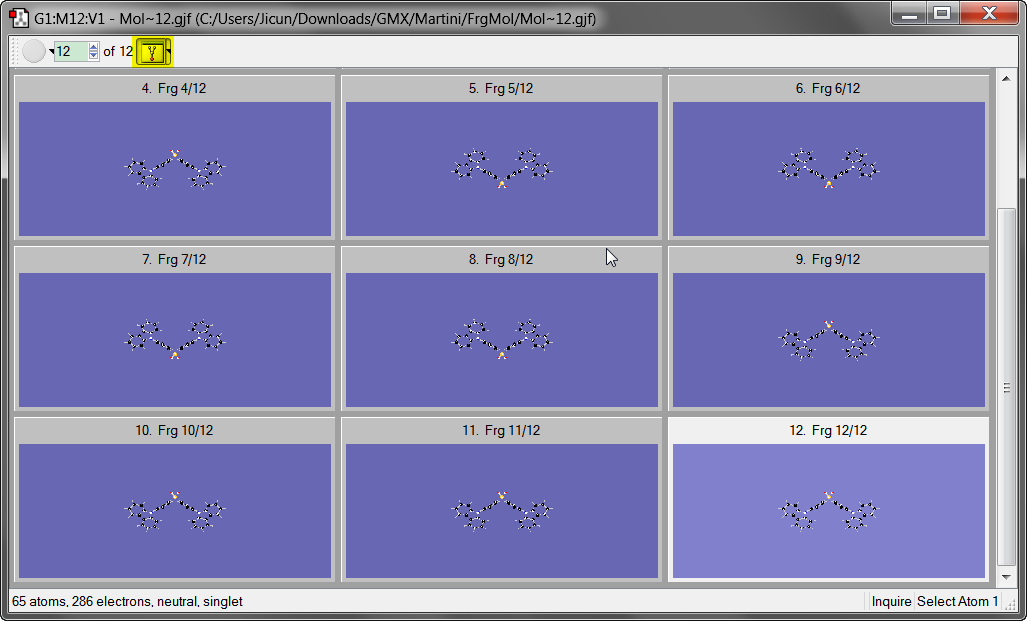
打开所有这些分子后, 点击Y形图标将所有12个分子排列好
菜单Edit | Connection打开连接编辑器, 在第一个分子中随便选择一个原子, 然后点击Enable Autofixing, 后面所有分子就会自动调整顺序.
要保存的话, 点击OK, 退出连接编辑器, 然后分别保存每个分子, 保存后的结构中原子的顺序就是对应好的了. 如果分子很多的话, 手动做起来也还是麻烦.
还要注意的是, 在连接编辑器中也可以手动指定每个原子的对应原子, 作法是左键选中第一个分子中的一个原子, 在其他分子中使用右键指定对应原子. 这样做比手动调整坐标顺序方便一些.
