- 2016-12-31 17:14:51 感谢 吴思晋 提供材料
氢键在分子动力学模拟结果的分析中占有重要地位, 因而大多数程序都带有氢键分析功能. 但需要注意的是, 不同程序中的氢键判定标准未必一致. 实际上, 氢键的判定有多种标准, 如能量准则, 电子结构准则, 几何准则等. 前两种准则计算量大, 不易实现, 因此大多数分析程序使用的都是几何准测. 即便同样是使用几何准测, 又有各种不同的方式, 如单纯的距离准则, 混合的距离-角度准则等. 其中的每种又可以细分. 具体的细节这里就不多细说了, 需要了解的话请参考论文: R. Kumar, J. R. Schmidt, J. L. Skinner; Hydrogen bonding definitions and dynamics in liquid water; 126(20):204107, 2007; 10.1063/1.2742385.
GROMACS和VMD是在分子动力学模拟中经常用到的程序, 它们都带有氢键分析功能, 所以在这里我们详细讨论下它们的氢键判定标准. 对其他程序所用标准, 也可进行同样的分析.
先给出氢键判定标准的几何准则所对应的结构图
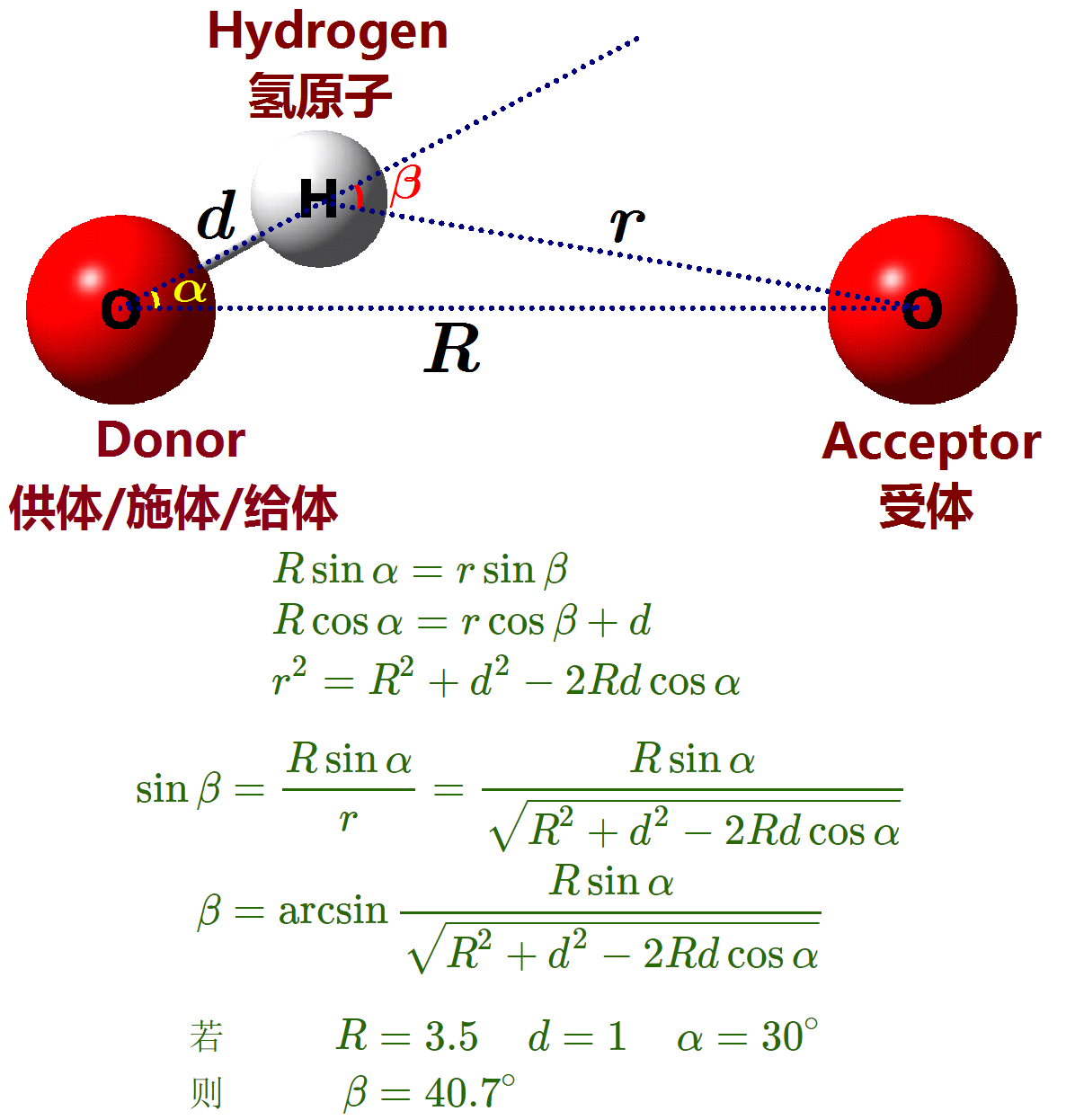
GROMACS的氢键判定
GROMACS的氢键分析工具为hbond, 其判定标准有两处说明: 手册8.12节和hbond文档.
GROMACS手册5.1.2版本8.12节说明
gmx hbond程序用于分析所有可能的施体D和受体A之间的氢键(HB). 分析时使用几何准则决定氢键的存在与否, 参看图 8.8.
$\alg
r &\le r_{HB} = 0.35 \ \text{nm}
\alpha &\le \alpha_{HB} = 30^{\circ}
\ealg$
参考值 $r_{HB}=0.35\ \text{nm}$ 对应于SPC水模型RDF的第一极小位置.
gmx hbond程序以下面的方式分析两组原子(它们必须相同或没有重叠)或指定的施体-氢-受体之间所有可能存在的氢键.
hbond文档5.1.2版本说明
gmx hbond用于计算和分析氢键. 氢键是由氢原子-施体-受体所成角度(0为扩展)的截断值与施体-受体之间距离(当使用-noda时为氢原子-受体距离)的截断值共同决定的. OH和NH被认作氢键施体, O总是作为氢键受体, N默认为受体, 但可以利用nitacc更改为施体. 哑的氢原子被假定为与前面的第一个非氢原子相连.
你需要指定用于分析的两个组, 它们必须完全相同或者彼此之间无任何重叠. 程序会分析两组间形成的所有氢键.
控制选项
-a <real> 30: 角度截断值(度, 氢原子‐施体‐受体)-r <real> 0.35: 半径截断值(nm, X‐受体, 见下一选项)-[no]da yes: 使用施体‐受体距离(若为真)或者氢原子‐受体距离(若为假)
VMD的氢键判定
VMD中的氢键判定标准有三处说明, 两处在其手册中, 一处在其HBonds插件的说明.
VMD手册1.9.3版本6.1.4节 HBonds
The ‘HBonds’ representation will draw a dotted line between two atoms if there is a possible hydrogen bond between them. A possible hydrogen bond is defined by the following criteria:
Given an atom D with a hydrogen H bonded to it and an atom A which is not bonded to D, a hydrogen bond exists between A and H iff the distance ||D-A|| < dist and the angle D-H-A < ang, where ang and dist are user defined.
Only the selected atoms are searched, so both the donor and acceptor must be selected for the bond to be drawn. Also, you’ll note that the above doesn’t check the atom type of the donor or acceptor; the only criterion is if it already has or doesn’t have a hydrogen.
One downfall of the current implementation is that it does an $n^2$ search of the selected atoms so you probably don’t want to show all the HBonds of a very large structure. Look for performance improvements in future versions of VMD.
If you choose an HBonds representation but fail to see any hydrogen bonds, it may be because the default Angle Cutoff and Distance Cutoff criterion in VMD are too small, so you might want to try increasing the angle value from 20 to 30 degrees and the distance value from 3 to 4.
VMD手册1.9.3版本9.3.18节 measure
hbonds cutoff angle selection1 [selection2]: Find all hydrogen bonds in the given selection(s), using simple geometric criteria. Donor and acceptor must be within the cutoff distance, and the angle formed by the donor, hydrogen, and acceptor must be less than angle from 180 degrees. Only non-hydrogen atoms are considered in either selection. If both selection1 and selection2 are given, the selection1 is considered the donor and selection2 is considered the acceptor. If only one selection is given, all non-hydrogen atoms in the selection are considered as both donors and acceptors. The two selections must be from the same molecule. The function returns three lists; each element in each list corresponds to one hydrogen bond. The first list contains the indices of the donors, the second contains the indices of the acceptors, and the third contains the index of the hydrogen atom in the hydrogen bond.
Known Issue: The output of hbonds cannot be considered 100% accurate if the donor and acceptor selection share a common set of atoms.
VMD插件 HBonds Plugin, Version 1.2
The Hbonds plugin counts the number of hydrogen bonds formed throughout a trajectory. The search can be restricted to a single selection or between two distinct selections, as well as a frame range given by the user.
Criteria for the formation of a hydrogen bond
A hydrogen bond is formed between an atom with a hydrogen bonded to it (the donor, D) and another atom (the acceptor, A) provided that the distance D-A is less than the cut-off distance (default 3.0 Angstroms) and the angle D-H-A is less than the cut-off angle (default 20 degrees).
总结说明
-
不同氢键判定标准得到的氢键数目不会完全相同, 因此, 在论文中报告氢键数目时必须说明使用的是哪种判定标准和具体的准则.
-
GROMACS氢键判定标准使用的几何准则默认为 $R-\a(3.5-30)$ 准则, 但也可以使用
-noda选项修改为 $r-\a$ 准则. -
VMD氢键判定标准使用的几何准则默认为 $R-\b(3.0-20)$ 准则.
-
对比GROMACS和VMD的氢键判定准则, 会发现VMD的默认准则远比GROMACS的严格, 因此, 使用默认准则计算时, 对于液态水分子, VMD得到的水分子形成的平均氢键数目会小于GROMACS的值, 而GROMACS的得到的值才比较符合大家的经验, 每个水分子的平均氢键数大约为3.5. 因此, 在实际应用中, 很少有人直接使用VMD的默认准则, 而是会使用更加宽泛的准则, 如 $R-\b(3.5-30)$, $R-\b(3.5-35)$. 但即便这个准则仍不等同于GROMACS的 $R-\a(3.5-30)$ 准则. 根据前面图中的几何关系, 可以看到VMD的 $R-\b(3.5-40)$ 准则才大致与GROMACS的 $R-\a(3.5-30)$ 准则等同. 但由于不存在准确的换算关系, 这两种准则是没有办法完全一致的.
-
需要注意的是, VMD计算氢键时, 并不会考虑周期性边界条件PBC, 因而对于一些小分子溶液的模拟, 使用VMD分析氢键时要加倍小心. 你可能发现即便你将VMD的准则放宽到 $R-\b(3.5-40)$, 得到的结果与GROMACS相比可能仍会偏小. 在这种情况下, 你要确定这是否是由于PBC引起的, 而不是盲目地继续放宽VMD的准则到 $R-\b(4.0-40)$. 有些人发现VMD得到的结果偏小就放宽准则, 直到得到自己觉得合适的值, 这种做法严格来说是错误的.
-
如果你不得不使用VMD的
measure hbonds进行氢键分析, 而又必须处理PBC的问题, 那解决方式可能有两种, 一是多次使用PBC转换和measure hbonds直到得到的氢键数目不变, 二是将边界处的分子排除掉, 在统计氢键时忽略它们. 这两种方法都是权宜之计, 最根本的还是要自己写代码分析, 或者让VMD支持PBC下的氢键分析.
