- 2017-01-11 22:28:27
我一直不喜欢用VMD, 除了其界面复杂, 鼠标操作模式老旧以外, 初始的显示模式也很简陋. 话虽这样说, 但有时还不得不用, 因为目前看来使用VMD查看轨迹还是最高效的. 毕竟, 人家名字就是 VVVVVVVV MD.
其实, 我们可以自定义VMD的初始显示模式, 让它变得友好一些. 这些设置可以放在和vmd.exe相同的目录下, 文件名称一般是vmd.rc. 这是VMD用于初始化的脚本, 我们可以在里面加上自己的tcl脚本, 满足自己的需要.
下面是我使用的VMD初始化脚本, 和原先的相比, 主要改动有三点:
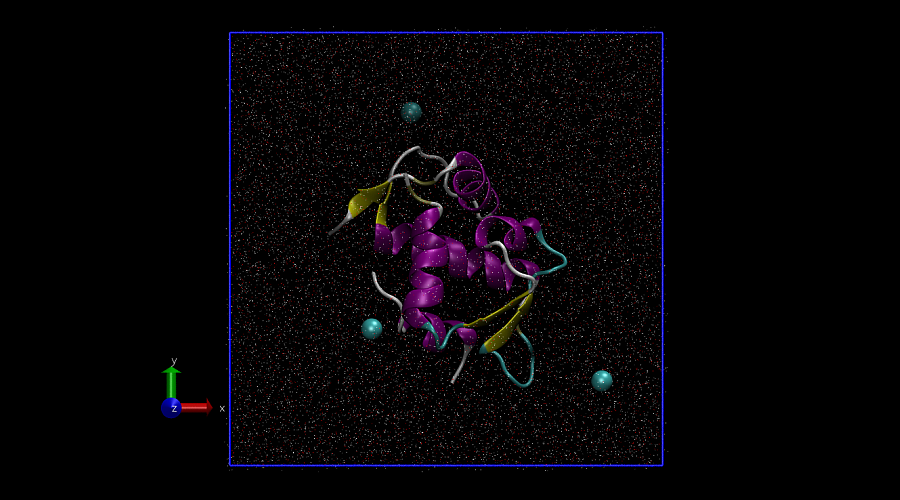
- 使用正视图而不是默认的透视图
- 有盒子大小信息时, 自动显示盒子
-
增加了一段tcl脚本, 根据分子情况决定其显示模式
- 蛋白质: newcartoon, 根据stucture着色
- 核酸: newribbons, 根据segname着色
- 水分子: 个数300以下, lines; 300以上, points
- 其他非标准残基, 配体: 原子个数500以下, CPK; 500以上, lines
- 不成键的原子(主要是离子和单个的氧原子): VDW
在这种模式下, 载入结构后看起来很清晰, 大多时候也无须自己再手动添加不同的表示模式, 方便不少.
| vmd.rc | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 | ############################################################################
#cr
#cr (C) Copyright 1995-2007 The Board of Trustees of the
#cr University of Illinois
#cr All Rights Reserved
#cr
############################################################################
############################################################################
# RCS INFORMATION:
#
# $RCSfile: .vmdrc,v $
# $Author: johns $ $Locker: $ $State: Exp $
# $Revision: 1.9 $ $Date: 2007/01/12 20:12:44 $
#
############################################################################
# DESCRIPTION:
#
# VMD startup script. The commands here are executed as soon as VMD starts up
############################################################################
# turn on lights 0 and 1
light 0 on
light 1 on
light 2 off
light 3 off
# position the stage and axes
axes location lowerleft
stage location off
# position and turn on menus
menu main move 5 225
menu display move 395 30
menu graphics move 395 500
menu color move 125 225
menu files move 125 325
menu labels move 125 525
menu render move 125 525
menu main on
#menu display on
#menu graphics on
#menu color on
#menu labels on
#menu render on
#menu files on
# start the scene a-rockin'
# rock y by 1
# User's default values for newly loaded molecules/reps, etc
# mol default color {colorID 5}
# mol default style {NewCartoon 0.300000 6.000000 4.100000 0}
# mol default selection {name CA}
# mol default material {Transparent}
display projection orthographic
after idle {
foreach molid [molinfo list] {
mol delrep 0 $molid
set sel [atomselect $molid {protein}]
if {[$sel num]} {
mol selection {protein}
mol representation newcartoon
mol addrep $molid
mol modcolor 0 $molid structure
mol selection {protein and (name CA or not backbone)}
mol representation lines
mol addrep $molid
mol modcolor 1 $molid resid
}
set sel [atomselect $molid {nucleic}]
if {[$sel num]} {
mol selection {nucleic}
mol representation newribbons
mol addrep $molid
mol modcolor 0 $molid resid
}
set sel [atomselect $molid {water}]
set num [$sel num]
if {$num} {
mol selection {water}
if {$num <900 } {
mol representation Lines
} else {
mol representation Points
}
mol addrep $molid
}
set txt "not(numbonds=0 or water or resname DA or resname DG or resname DT or resname DC or resname ALA or resname ARG or resname ASN or resname ASP or resname CYS or resname GLN or resname GLU or resname GLY or resname HIS or resname HOH or resname ILE or resname LEU or resname LYS or resname MET or resname PHE or resname PRO or resname SER or resname THR or resname TRP or resname TYR or resname VAL)"
set sel [atomselect $molid "$txt"]
set num [$sel num]
if {$num} {
if {$num <500 } {
mol representation CPK
} else {
mol representation Lines
}
mol selection "$txt"
mol addrep $molid
}
set txt "numbonds=0"
set sel [atomselect $molid "$txt"]
if {[$sel num]} {
mol selection "$txt"
mol representation VDW
mol addrep $molid
}
}
pbc box
}
|
当然, 这个初始化脚本只有在命令行中启动VMD时才会自动加载的. 如果你是使用界面来载入分子, 那它是不会起作用的. 建议大家还是习惯使用命令行来启动VMD吧, 毕竟可以节省不少时间, 且有助于预防鼠标手.
命令行启动VMD加载分子没有你想象得那么高深. 对Windows 7及以后的系统, 将vmd.exe所在的目录添加到Path环境变量后, 到分子结构文件所在的目录下, 不要选中任何东西, 按住Shift键, 在界面空白处鼠标右键, 选择在此处打开命令窗口打开CMD, 然后在其中输入vmd file.pdb就可以直接打开file.pdb了. 查看GROMACS的轨迹文件, 则可以使用vmd conf.gro traj.trr或者vmd conf.gro traj.xtc. 其他格式的文件, 也是类似.
评论
- 2017-02-26 11:22:49
.很好用! -
2017-02-26 19:00:48
Jerkwin有用就好. - 2017-04-14 10:07:04
路易斯李老师,这个脚本在linux下可以用吗?没找到vmd.rc的文件位置。不知道应该放在哪里? - 2017-04-14 21:22:39
JerkwinLinux下VMD的目录下会有个.vmdrc, 用这段脚本替换原来的就可以了. - 2017-04-17 10:36:28
路易斯谢谢李老师
