- 原始文档QM/MM SIMULATION
- 2017-02-14 15:20:45 翻译: 郑伟中; 校对: 李继存
- 2017-10-28 16:55:24 增补: 陈国俊
此链接提供了一个很好的QM/MM教程, 但它有点复杂, 而且缺少一些细节以致初学者很难重复整个教程. 因此, 我们利用一个简单的体系来帮助初学者一步一步地进行一个QM/MM模拟.
【陈国俊】 首先在教程前说一点闲话, 我从2016年寒假开始折腾GROMACS的QM/MM, 中间断断续续有小半年时间, 期间也看了用DFTB3进行QM/MM模拟的方法, 但是当时怎么编译安装都没能成功, 不是PLUMED出错, 就是GROMACS出错, 所以最后无奈放弃. 在其他量化软件里面, 除了GAUSSIAN我一个都不熟, 所以就开始折腾GAUSSIAN+GROMACS的QMMM方法. 一开始除了GROMACS官网里的那点简述和李老师及其他热心群友翻译的几篇教程, 可供参考的资料少之又少, 完全就是照葫芦画瓢, 犯了很多低级的错误, 比如新老版本参数不分等. 后来又开始仔细读手册, 在群里向群友们请教. 上次在北京参加李老师的培训班, 又和李老师讨论了这个事情, 最后终于调试成功, 所以整理了一下我的整个运行流程, 希望能给用得到的朋友带来一点帮助.
二点经验很重要:
- 手册不要离手, 出错了, 回去翻翻手册会找到很多新的启示;
- 要对拓扑文件有充分的了解, 这样才可以正确地编写相应的设置.
0. 准备工作
在开始此教程之前, 我们假定:
- GROMACS和GAUSSIAN软件已正确安装(本教程使用的GROMACS为2016.4版本)
- 对GROMACS操作有一定的基础
- 阅读GROMACS官网的QM/MM文档或其翻译
- 了解GROMACSQM/MM实现方法的原理, 并按其中的说明设置好相应的环境变量.
- 本教程在Windows平台上完成, 对Linux平台也是一样的流程.
- 可以下载本教程用的文件
【陈国俊】 对于使用GAUSSIAN的情况, 本教程采用的是使用外挂脚本的方法, 所以不需要修改GAUSSIAN的源码. 我用的是李老师整理过的脚本. 使用这个脚本方便的地方在于, 你可以使用任意的QM方法, 而不仅仅限于GROMACS支持的那几种. 比如你可以使用半经验的PM6方法, 如果要考虑弱的色散相互作用, 你可以在自己准备的文件里添加关键词
EmpiricalDispersion=GD3.
1. 构建模拟体系, 创建gro和top文件
随意构建一条小肽, 如ALA ALA ALA SER GLY ALA ALA ALA. 我们的目的是把SER和GLY作为QM区域进行QM/MM模拟. 小肽的结构文件命名为pep.pdb.
我们将使用AMBER99SB-ildn力场, 为获得拓扑文件, 运行下面的命令并选择AMBER99SB-ildn力场以及TIP3P水模型:
gmx pdb2gmx -f pep.pdb -o pep.gro -p topol.top -ignh
然后添加盒子:
gmx editconf -f pep.gro -o conf.gro -d 1
这样我们获得了三个文件: 结构文件conf.gro, 拓扑文件topol.top和位置限制文件porse.itp.
2. 添加连接原子(LA, Link Atom)信息, 修改gro和top文件
这里我们不解释为什么要在QM/MM模拟中引入连接原子, 你自己很容易找到答案.
下面的图片展示了我们如何把整个体系分为QM部分和MM部分.
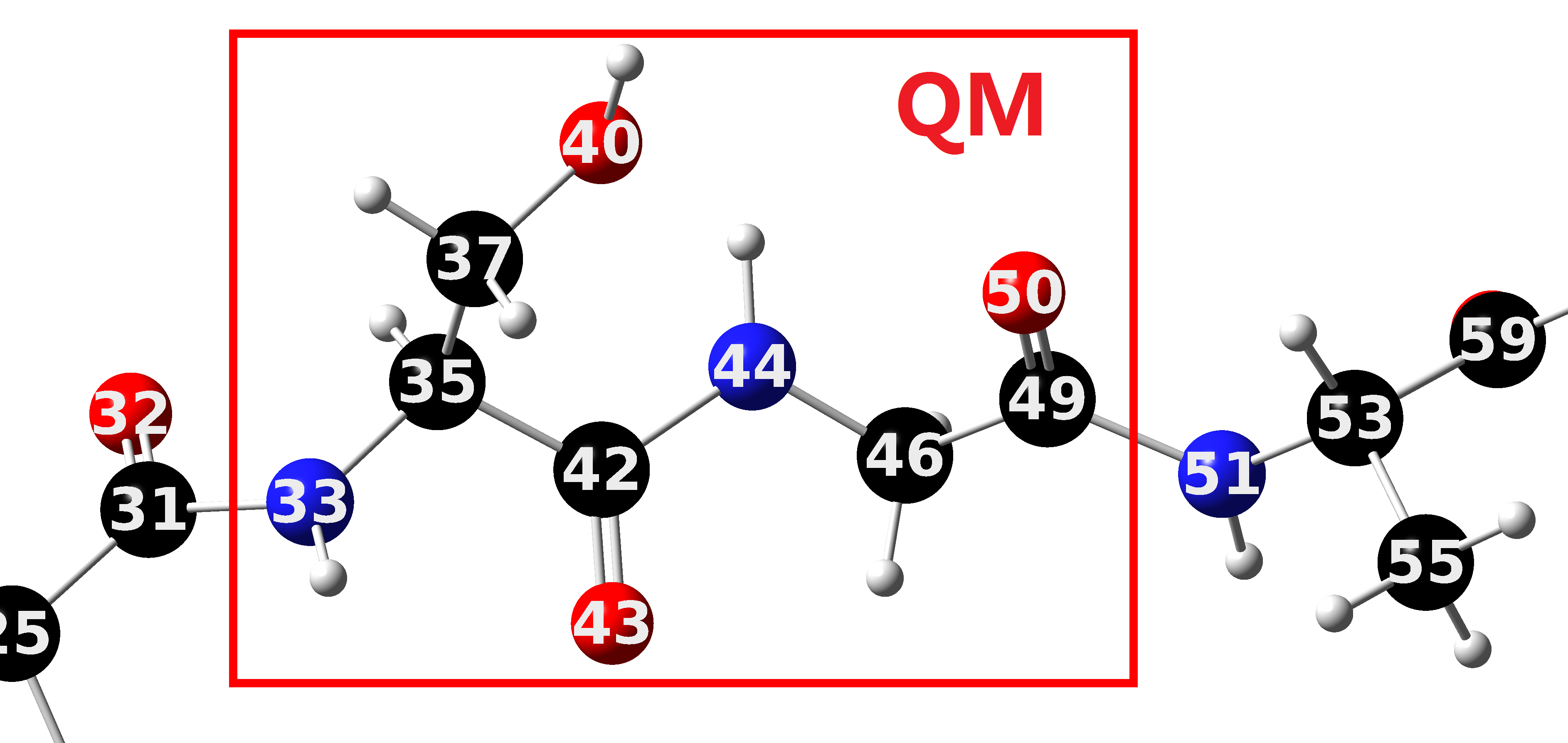
确定了QM区域以后, 我们需要将连接原子添加到结构文件conf.gro, 并在拓扑文件topol.top中定义连接原子的原子类型及其参数, 同时修改与其相关的成键作用参数.
修改结构文件conf.gro
如上图所示, 由于我们划分QM区域与MM区域时破坏了原先的成键, 所以我们要在QM和MM区域的边界处中添加两个连接原子(假定其名称为LA), 因此体系的总原子数应比原来增加2. 打开结构文件conf.gro, 首先将第二行中体系的总原子数由81改为83. 然后我们还要添加这两个LA原子的坐标.
值得注意的是, 如果你的体系中有多个分子, 那么添加LA时就要注意添加的位置. 原则上要添加到LA所属分子的最后一个原子的后面. 由于我们的体系中只有一个分子, 所以直接将LA添加到最后一个原子的后面即可. 但要注意格式与前面的一致.
| conf.gro | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 | GROningen Mixture of Alchemy and Childrens' Stories
83
1ALA N 1 1.122 1.070 1.170
1ALA H1 2 1.149 1.019 1.088
1ALA H2 3 1.149 1.019 1.252
1ALA H3 4 1.022 1.082 1.170
1ALA CA 5 1.186 1.200 1.170
1ALA HA 6 1.153 1.243 1.086
1ALA CB 7 1.147 1.280 1.293
1ALA HB1 8 1.192 1.369 1.291
1ALA HB2 9 1.048 1.294 1.294
1ALA HB3 10 1.174 1.231 1.376
1ALA C 11 1.338 1.186 1.170
1ALA O 12 1.390 1.074 1.170
....(略)
8ALA N 71 3.279 2.225 1.834
8ALA H 72 3.298 2.269 1.746
8ALA CA 73 3.384 2.212 1.933
8ALA HA 74 3.338 2.166 2.009
8ALA CB 75 3.501 2.130 1.880
8ALA HB1 76 3.571 2.123 1.950
8ALA HB2 77 3.469 2.039 1.854
8ALA HB3 78 3.539 2.176 1.799
8ALA C 79 3.437 2.348 1.975
8ALA OC1 80 3.510 2.354 2.044
8ALA OC2 81 3.392 2.451 1.925
9QMM LA1 82 0.000 0.000 0.000
9QMM LA2 83 0.000 0.000 0.000
4.54900 3.43200 3.08000
|
上面添加到两行内容中, 第一列是残基名称, 并不重要, 可以随便用其他名字来替代9QMM; 第二列是原子名称, 我们使用前面指定的名称LA. 这个名称也可以随意写, 只要拓扑文件中指向的原子类型与定义的LA原子类型相同即可. 但与拓扑文件中不一致时grompp会出现警告; 第三列是原子序号, 根据前面的编号继续即可(实际上也可以随意写, 只要总原子数目对即可); 最后三列是原子的坐标, 我们可以全部指定为零, 因为LA属于虚拟位点, 对于虚拟位点的坐标, GROMACS在运行时不会使用结构文件中的值, 而是根据拓扑文件中的信息和结构文件中定义虚拟位点的原子的坐标自动计算. 更具体的说明见后面修改拓扑文件部分.
如果你不喜欢使用零作为LA的坐标, 那么可参考下面的方法来确定LA的坐标(也可参考此链接的adding link atoms部分.
残基3ALA的C31原子和残基4SER的N33原子之间的连接原子应位于这两个原子之间, 它与N33原子之间的距离应正比于C31和N33原子之间的键长. 由于进行QM/MM计算时我们将连接原子视为氢原子, 它距所连氮原子的的距离并不是固定值, 而是根据原结构中的C-N距离来确定. 通常情况下C(=O)-N键长为0.152 nm, N-H键长为0.101 nm, 其比例 $k=0.101/0.152=0.664$. 这样将C31和N33之间的实际键长乘以0.664就可以得到N33和LA1之间的距离. 再根据C31和N33原子的坐标, 就可以计算出LA1的坐标了. 用数学公式表示的话, 可写为 $\vec R_\text{LA1}=\vec R_\text{N33}+k\vec R_\text{N33-C31}$.
同理, C49和N51之间的LA2原子的坐标也可以计算出来, 只不过LA2是连接在碳原子上的. 通常情况下C(=O)-N键长为0.152 nm, C-H键长为0.108 nm, 其比例为0.108/0.153=0.706.
修改拓扑文件topol.top
首先需要向力场中添加LA的原子类型. 我们既可以将LA类型直接添加到GROMACS自带的力场文件中(或复制一套放于工作目录下, 更稳妥), 也可以将其添加到我们前面得到的topol.top中. 一般建议使用后者, 因为使用前者容易污染GROMACS自带的力场文件. 为此, 我们需要修改前面得到的拓扑文件topol.top. 首先添加LA的原子类型, 将下面的内容添加到#include(力场头文件)语句的下面. 注意, 添加到位置非常重要, 一定要保证[ atomtypes ]位于展开内容之后[ defaults ]的后面.
| topol.top | |
|---|---|
1 2 | [ atomtypes ]
LA 1 0 0 A 0 0 ; LA for QM/MM
|
添加了LA的原子类型后, 还应在拓扑文件中分子类型部分添加LA以保持拓扑与结构文件一致. 首先, 我们在拓扑文件[ atoms ]部分的最后增加下列内容:
| topol.top | |
|---|---|
1 2 | 82 LA 9 QMM LA1 82 0.000 0
83 LA 10 QMM LA2 83 0.000 0
|
同样, 第一列是LA原子在所属分子类型中的编号, 第二列对应的力场中的原子类型(注意与原子名称的不同), 第三列是残基编号, 第四列是残基名称(可随意, 不影响结果), 第五列是原子名称(可随意, 但最好保持与结构文件中的一致, 否则grompp会警告. 注意与原子类型的意义不同), 第六列是原子序号, 然后是原子电荷(LA原子不带电, 所以为零), 最后一列是原子的质量(虚拟位点质量为零).
接下来我们需要修改分子的成键相互作用部分, 将QM和MM原子之间的成键类型改为5, 表示两个原子之间是无相互作用的连接. 具体来说, 就是[ bonds ]部分中, 改为31 33 5和49 51 5.
在一些教程中, 还会建议下面的修改, 但根据我的测试, 下面的修改对结果并无影响, 因此可以省略.
对于[ bonds ]部分, 我们需要把所有两个QM原子之间的成键类型改为5, 表示这些原子之间是无相互作用的连接.对于[ angle ]部分, 我们应该移除QM区域内的键角. 如果形成键角的三个原子中至少有两个原子属于QM原子, 那么我们就认为这个键角处于QM区域内, 应该被移除.对于[ dihedrals ]部分(正常或异常二面角), 我们应移除QM区域内的二面角. 如果形成二面角的四个原子中至少有三个原子属于QM原子, 那么我们就认为这个二面角处于QM区域内, 应该被移除.
最后, 我们需要在拓扑文件中定义LA原子所在虚拟位点. 所谓定义虚拟位点, 就是指定虚拟位点的约束条件. 在之后的模拟过程中, GROMACS会按照定义好的约束条件来更新虚拟位点的坐标. 我们之前说过, 在结构文件中添加LA原子时可以不用给出具体坐标, 简单指定为零即可, 就是因为GROMACS会自动计算.
在[ dihedrals ]后面添加[ virtual_site2 ](对应GROMACS老版本中的[ dummies2 ]. 注意添加的位置):
| topol.top | |
|---|---|
1 2 3 4 | [ virtual_sites2 ]
; Site from funct a
82 33 31 1 0.664
83 49 51 1 0.706
|
其中, site是虚拟位点在分子结构中的编号, from表示根据哪些原子计算虚拟位点. 在我们的示例中是根据31和33来计算LA1, 根据49和51来计算LA2. funct为计算虚拟位点的函数类型, 这里是1, a为约束参数, 对应于我们前面的计算值. 手册 4.7 虚拟相互作用位点对虚拟位点的设置有详细的介绍, 可以参考.
再添加[ constraints ]部分:
| topol.top | |
|---|---|
1 2 3 | [ constraints ]
33 31 2 0.152
49 51 2 0.152
|
这里的0.152是C, N间的键长,
到这里GROMACS文件全部修改完成了.
3. 能量最小化
在进行QM/MM模拟之前, 可以先进行一下简单的能量最小化, 用以检查前面的输入文件是否正确, 同时也可以得到虚拟位点的坐标.
gmx grompp -f grompp_em.mdp -c conf.gro -p topol.top -o em.tpr
gmx mdrun -v -deffnm em
完成后会得到能量最小化之后的结构em.gro. 用可视化软件查看下, LA是否处于正确的位置(如果发现显示的成键连接很乱, 不用管它, 只是显示的问题).
4. 创建索引文件
在运行QM/MM模拟时, GROMACS需要知道哪些原子应使用QM进行处理. 所以就需要我们指定处于QM区域内的原子, 并将其单独列为一组, 保存在索引文件中. 对于小体系, 可以直接手写索引文件. 对复杂点的体系使用gmx make_ndx可以轻松完成. 要注意的是, 在生成QM原子组时, 要把虚拟原子也包括进来, 否则的话, GAUSSIAN运行时会缺少连接原子, 构型不对, 得到错误的结果.
要创建QM原子组, 对我们的体系, 可使用下列命令:
gmx make_ndx -f em.gro -o index.ndx
> r 4 5 | r QMM
> name 14 QMatoms
> q
这样我们就得到了一个索引文件index.ndx. 其中包含了QM原子组QMatoms:
| index.ndx | |
|---|---|
1 2 3 | [ QMatoms ]
33 34 35 36 37 38 39 40 41 42 43 44 45 46 47
48 49 50 82 83
|
5. 创建运行参数文件
由于示例体系很简单, 也不考虑溶剂, 所以我们省略预平衡阶段, 直接进行MD模拟. 进行MD前, 需要首先设置好GAUSSIAN运行所需的环境变量. 然后在grompp.mdp文件中指定QM/MM选项, 选项中的参数取决于你的体系. grompp.mdp文件中的其他设置与通常模拟没有区别.
我们指定的QM/MM选项如下:
| grompp.mdp | |
|---|---|
1 2 3 4 5 6 7 8 | QMMM = yes ; QM/MM模拟开关打开
QMMM-grps = QMatoms ; QM原子所属组
QMmethod = B3LYP ; QM的理论方法
QMbasis = 6-31G* ; QM使用的基组
QMMMscheme = normal ; QM/MM的类型(只有一个QM组用normal即可)
QMcharge = 0 ; QM原子的净电荷
QMmult = 1 ; QM原子的自旋多重度
SAsteps = 0 ; GMX2016.4需要此选项
|
这里需要额外说明的是, GROMACS支持的QMmethod只有几种, 如果换用其他基组grompp时还会报错, 所以我们随意指定GROMACS支持的选项即可. 我们在这里定义的QMmethod和QMbasis实际上并不会用到, 实际运行时使用的设置来自我们提前准备好的GAUSSIAN模板文件_Gkwd.gjf. 这样我们才可以使用GAUSSIAN支持的任意理论方法和基组. 根据GROMACS QM/MM教程2:编译设置及简单运行的说明准备_Gkwd.gjf. 可以参考如下示例:
| _Gkwd.gjf | |
|---|---|
1 2 3 4 | %chk=input
#T HF NoSymm
Force Punch=Derivatives
Charge Prop=(Field, Read)
|
6. 运行模拟
到目前为止, 我们已经准备好了运行模拟所需的所有文件. 输入下面的命令进行模拟:
gmx grompp -f grompp.mdp -c em.gro -p topol.top -n index.ndx -o nvt.tpr -maxwarn 3
gmx mdrun -deffnm nvt
如果不出意外, 你应该看到类似下面的输出:
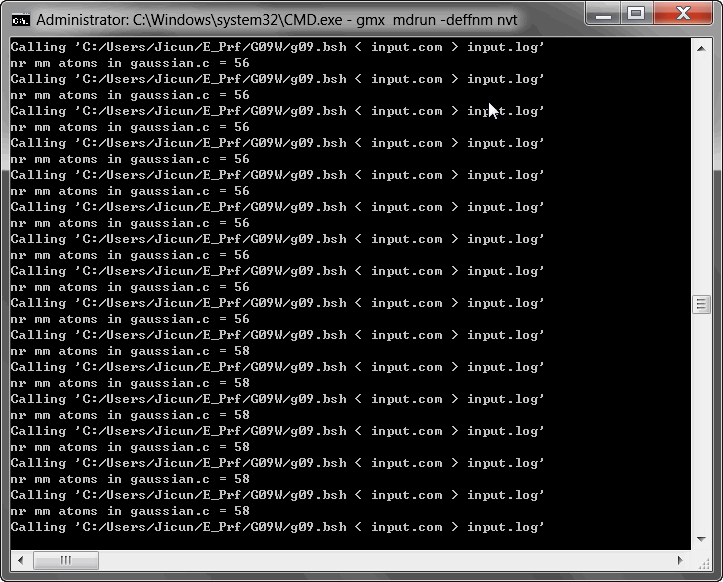
并且轨迹文件和日志文件也在更新. 这说明模拟已经成功地运行了. 祝贺你. 不过也别高兴的太早, 因为QM/MM运行过程中经常会出现构型问题, 以及GAUSSIAN Link 502自洽迭代不收敛的错误, 你可能还需要修改mdp选项, 并增加一些GAUSSIAN控制SCF收敛的选项才能顺利地模拟下去. 由于本教程仅为了示例如何运行一个QM/MM模拟, 所以我们就不再继续讨论这些细节了. 此外, 要进行一次有意义的模拟, 你需要学习更多分子模拟的知识, 这也超出了此教程的范围.
7. 结语
【陈国俊】 在结束部分, 还有点闲话想说一下. 根据我的测试, 这种QM/MM方法计算速度极慢, 只适用于小体系的短时间模拟. 即便对于本教程使用的这样小的示例体系, 在我的电脑上(i7-6700k), 使用PM6D3这种半经验方法模拟100 ps都需要两三个小时, 而使用B3LYP/6-31G*这种并非很高精度的方法时测试的速度是44 day/ns. 我估计运行如此之慢的原因一是因为GROMACS不能并行, 二是GAUSSIAN本身运算的速度就很慢, 三是这两者中间还需要一个脚本进行数据交换, 其速度可想而知. 如果想做大体系长时间的模拟, 还是趁早换其他的门路, 不要在这折腾浪费时间了.
