- 原始文档: Ross Walker and Sishi Tang, TUTORIAL B4: Simulating a pharmaceutical compound using antechamber and the Generalized Amber Force Field
- 2018-01-03 15:52:14 翻译: 李睿; 修订: 李继存
antechamber是AMBER自带的一组工具, 可用于准备有机分子的prep输入文件, LEaP可以读取这种输入文件并将其用于创建prmtop和inpcrd文件. antechamber程序可以与GAFF(general AMBER force field)联合使用, 非常适于设置涉及有机药物分子或其他有机分子的模拟. 在本教程中, 我们将使用antechamber为BMS的HIV逆转录酶(HIV-RT)抑制剂Sustiva(efavirenz, 中文名依法韦仑)创建leap输入文件, 然后我们将对Sustiva与HIV-RT的复合物进行模拟.
antechamber教程: 使用antechamber和GAFF力场创建LEaP输入文件用于模拟Sustiva(efavirenz)-RT复合物
- 编写: Ross Walker and Sishi Tang
- 2018-01-03 15:52:14 翻译: 李睿; 修订: 李继存
简介
在本教程中, 我们将使用AmberTools自带的Antechamber软件包, 生成蛋白-配体复合物的prmtop和inpcrd文件, 并进行短时间的GB模拟.
Antechamber设计与通用AMBER力场(GAFF, general AMBER force field)一起使用[1]. 这个力场是特别设计的, 涵盖了大多数药物分子, 且与传统AMBER力场兼容. 这样, 在模拟中可以将这两种力场混合起来, 共同用于处理蛋白和药物分子形成的复合物. 就像传统的AMBER力场一样, GAFF使用简单的简谐势函数来描述键和键角. 但与针对蛋白质和DNA的传统AMBER力场不同的是, GAFF使用的原子类型通用得多, 因此可以涵盖大部分有机分子. 目前的GAFF力场实现中包括了基本的原子类型和特殊的原子类型. 电荷的计算方法可以是HF/6-31G* RESP[2]或AM1-BCC[3].
设计时, GAFF力场是一种完整的力场(因此缺失的参数非常少). 它几乎涵盖了所有由C, N, O, S, P, H, F, Cl, Br和I等元素组成的有机化合物. 此外, 因为GAFF完全兼容AMBER大分子力场, 所以它应该能够成为一个有用的分子力学工具, 用于进行理性的药物设计, 尤其是在结合自由能计算和分子对接研究方面.
Antechamber工具集设计用来快速地生成拓扑文件, 用于AMBER模拟程序. 当你需要自动筛选大量化合物时, 这个工具十分有用. 使用GAFF时, antechamber可用于自动计算电荷和原子类型, 这样就可以在shell脚本中使用它们处理大量的化合物, 大大加快了处理速度. 然而, 就像其他的自动化计算程序一样, Antechamber并不完美. 如果你计划专注于单个体系的模拟, 就应该考虑手动指定原子类型, 并小心确证模拟的方方面面. 使用Antechamber时, 你可能解决以下问题:
- 自动识别化学键以及原子类型
- 判断原子等价性
- 生成残基拓扑文件
- 发现缺失的力场参数并提供合理的建议值
然而, 请牢记Antechamber并不能替代对体系的严格核查. 你始终都应该仔细检查Antechamber指定的原子类型, 并自己验证其选择的合理性. 在使用科学软件时, 任何”黑箱”式的操作方式都是不可取的!
在本教程中我们将使用Antechamber工具集和LEaP来创建处方药Sustiva(Efavirenz, 依法韦仑)的拓扑文件和坐标文件. 依法韦仑是人免疫缺陷病毒类型1(HIV-1, 艾滋病病毒)的特异性非核苷类逆转录酶(RT, reverse transcriptase)抑制剂, 由Bristol Myers Squibb公司市场化, 用于控制人类HIV感染的过程. Sustiva的化学名称为(S)-6-chloro-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one, 化学式为C14H9ClF3NO2, 二维结构如下:
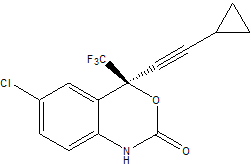
sustiva.pdb是Sustiva的3D结构, 我们将根据它来构建拓扑和坐标文件. 这个文件是从RT-sustiva复合物的PDB文件(PDB编号: IFKO)中抽取出来的. PDB文件中与Sustiva的坐标相应的残基名称为EFZ(代表Efavirenz).
首先使用VMD打开sustiva.pdb查看一下其结构.
我们将使用Antechamber指定这个分子中每个原子的原子类型, 并计算每个原子上的电荷. Antechamber是Antechamber工具集中最重要程序, 可以进行多种文件类型的转换并指定换原子类型以及原子电荷. 根据输入文件, Antechamber执行下列程序(AmberTools中包含了所有这些程序): sqm, atomtype, amlbcc, bondtype, espgen, respgen和prepgen. 它还会生成一系列中间文件(文件名全部为大写字母).
创建Sustiva的参数和坐标文件
首先, 使用reduce程序为PDB文件添加所有的氢原子:
reduce sustiva.pdb > sustiva_h.pdb
加氢后的sustiva坐标如下sustiva_h.pdb.
为了与PDB文件的名称保持一致, 我们将PDB文件中的残基名称EFZ改为SUS, 并将其另存为新的PDB文件sustiva_new.pdb.
现在使用antechamber来处理新得到的sustiva的PDB文件. 为了能够在LEaP中定义新的单元, 我们需要创建mol2文件, 简单地运行如下命令即可:
antechamber -i sustiva_new.pdb -fi pdb -o sustiva.mol2 -fo mol2 -c bcc -s 2
上面命令的选项说明如下:
-i sustiva_new.pdb: 指定输入3D结构文件的名称-fi pdb: 指定输入文件的格式为PDB(我们也可以使用任何其他支持的文件格式, 包括Gaussian的Z矩阵[gzmat], Gaussian输出文件[gout], MDL[mdl], amber重启[rst], Sybyl Mol2[mol2])-o sustiva.mol2: 指定输出文件的名称-fo mol2: 说明输出文件的类型为Tripos Mol2格式(这是LEaP支持的内部格式)-c bcc: 指示antechamber使用AM1-BCC电荷模型来计算原子上的电荷-s 2: 指定antechamber程序提供的状态信息的冗长度. 我们选择提供更多信息(2)
执行上面的命令, 屏幕输出应类似如下:
Running: /usr/local/amber10/bin/bondtype -j full -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac
Running: /usr/local/amber10/bin/atomtype -i ANTECHAMBER_AC.AC0 -o ANTECHAMBER_AC.AC -p gaff
Total number of electrons: 160; net charge: 0
Running: /usr/local/amber10/bin/mopac.sh
Running: /usr/local/amber10/bin/am1bcc -i ANTECHAMBER_AM1BCC_PRE.AC -o ANTECHAMBER_AM1BCC.AC -f ac
-p /usr/local/amber10/dat/antechamber/BCCPARM.DAT -s 2 -j 1
Running: /usr/local/amber10/bin/atomtype -f ac -p bcc -o ANTECHAMBER_AM1BCC.AC -i ANTECHAMBER_AM1BCC_PRE.AC
在工作目录下会产生一系列的文件:
ANTECHAMBER_AC.AC ANTECHAMBER_AM1BCC_PRE.AC ATOMTYPE.INF mopac.out sustiva.mol2
ANTECHAMBER_AC.AC0 ANTECHAMBER_BOND_TYPE.AC divcon.pdb mopac.pdb sustiva.pdb
ANTECHAMBER_AM1BCC.AC ANTECHAMBER_BOND_TYPE.AC mopac.in
名称全部大写的所有文件是antechamber计算时使用的中间文件, 并不重要也不需要保存, 你可以放心地删除它们. 程序默认没有删除这些文件是因为有些情况下计算可能出错, 这时可利用这些文件来排查错误. mopac.in和mopac.out文件是mopac输入与mopac输出文件, 用于antechamber计算电荷. 除了验证mopac计算是否成功, mopac文件在这里不需要.
我们之所以首先运行antechamber, 就是为了得到我们真正需要的sustiva.mol2文件. 这个文件中包含了sustiva 残基的定义, 包括所有的电荷信息以及原子类型. 我们可以将它载入LEaP, 用以创建prmtop以及inpcrd文件. 让我们来快速地浏览一下文件的内容:
| sustiva.mol2 | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | @<TRIPOS>MOLECULE
SUS
30 32 1 0 0
SMALL
bcc
@<TRIPOS>ATOM
1 CL -4.6850 -32.7250 25.2220 cl 999 SUS -0.073100
2 F1 -0.7550 -36.6320 25.6970 f 999 SUS -0.231400
3 F2 1.0780 -37.0430 24.6720 f 999 SUS -0.221400
4 F3 -0.7840 -37.1770 23.6260 f 999 SUS -0.216800
5 O1 1.5240 -34.9340 20.9100 o 999 SUS -0.573600
6 O2 0.9890 -34.8800 23.0580 os 999 SUS -0.371900
7 N -0.6810 -34.9710 21.4340 n 999 SUS -0.459500
8 C1 -1.6620 -34.4140 22.3130 ca 999 SUS 0.084700
9 C2 -2.9150 -33.9470 21.8430 ca 999 SUS -0.167000
10 C3 -3.8380 -33.4230 22.7710 ca 999 SUS -0.069500
11 C4 -3.5330 -33.3730 24.1190 ca 999 SUS -0.025500
12 C5 -2.3100 -33.8290 24.5930 ca 999 SUS -0.040200
|
可以看到, 这个文件中包含了sustiva分子的三维结构, 每个原子的原子序号(第一列), 原子名称(第二列), 原子类型(第六列)以及每个原子上的电荷(最后一列). 在文件的最后还指定了成键信息. 但是这个文件并不包含任何参数. GAFF的所有参数都是在$AMBERHOME/dat/leap/parm/gaff.dat中定义的. 此外, 你还应该注意到, 所有GAFF原子类型的名称都以小写字母表示. 这是为了保证GAFF力场独立于大分子的AMBER力场. 所有传统AMBER力场都使用大写字母表示原子类型. 这样就可以在一次模拟中同时使用GAFF和传统力场, 而不必担心混淆.
虽然参数文件中定义了大多数键, 键角, 二面角参数的可能组合, 但我们的分子仍可能包含了未知原子类型的组合, 其相应的键, 键角, 二面角参数不存在. 如果是这样的话, 在使用LEaP创建prmtop和inpcrd文件之前, 我们必须指定这些缺失的参数.
我们可以使用parmchk2检查mol2文件, 看是否存在缺失参数:
parmchk2 -i sustiva.mol2 -f mol2 -o sustiva.frcmod
# 如果使用parmchk2报错, 改用parmchk试试
执行上述命令后会生成sustiva.frcmod文件. 这是一个参数文件, 能够载入LEaP中用于添加缺失的参数, 这样就能包含所有缺失的参数. 如果可以, antechamber会根据对相似参数的类比补充缺失参数. 在模拟之前, 你应该仔细检查这些参数. 如果antechamber不能经验地计算一些参数的值, 也无法通过类比获得它们, antechamber会为这些参数指定它认为合理的值, 或者是插入占位空间(所有参数为0), 并返回ATTN: needs revision的信息. 在这种情况下, 你必须自己手动为这些参数指定合理的值. 随着GAFF的发展, 希望缺失参数的数目可以越来越少. 让我们看看frcmod文件的内容:
| sustiva.frcmod | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | remark goes here
MASS
BOND
ANGLE
ca-c3-c1 64.784 110.735 Calculated with empirical approach
c1-c1-cx 56.400 177.990 same as c1-c1-c3
c1-cx-hc 48.300 109.750 same as c1-c3-hc
c1-cx-cx 64.200 111.590 same as c1-c3-c3
DIHE
IMPROPER
ca-ca-ca-ha 1.1 180.0 2.0 General improper torsional angle (2 general atom types)
n -o -c -os 10.5 180.0 2.0 General improper torsional angle (2 general atom types)
c -ca-n -hn 1.1 180.0 2.0 General improper torsional angle (2 general atom types)
ca-ca-ca-n 1.1 180.0 2.0 Using default value
NONBON
|
我们可以看到, 一共缺失了4个键角参数和4个反常二面角参数. 考虑到本教程的目的, 我们直接假定antechamber建议的参数可以接受, 无须修改. 理想情况下, 你应该实际测试这些参数(例如通过与ab initio计算进行比较)以保证其合理性. 如果你看到任何列出的带有ATTN: needs revision注释的参数, 都意味着antechamber无法确定合适的参数, 在模拟之前你必须手动提供这些参数的值. 默认情况下antechamber会将这些参数设置为0.
现在我们已经准备好了将sustiva载入LEaP作为一个单元的所有工作, 只需要运行tleap保证GAFF力场能够使用即可.
tleap -f oldff/leaprc.ff99SB
tleap出现并运行后, 我们还需要保证tleap可以调用GAFF力场, $AMBERHOME/dat/leap/cmd/有一个脚本可以帮我们完成这件事. 我们可以使用下面的命令将其载入tleap:
source leaprc.gaff
现在我们的tleap终端看起来应该类似如下所示:
Welcome to LEaP!
(no leaprc in search path)
Sourcing: /usr/local/amber10/dat/leap/cmd/oldff/leaprc.ff99SB
Log file: ./leap.log
Loading parameters: /usr/local/amber10/dat/leap/parm/parm99.dat
Reading title:
PARM99 for DNA,RNA,AA, organic molecules, TIP3P wat. Polariz.& LP incl.02/04/99
Loading parameters: /usr/local/amber10/dat/leap/parm/frcmod.ff99SB
Reading force field modification type file (frcmod)
Reading title:
Modification/update of parm99.dat (Hornak & Simmerling)
Loading library: /usr/local/amber10/dat/leap/lib/all_nucleic94.lib
Loading library: /usr/local/amber10/dat/leap/lib/all_amino94.lib
Loading library: /usr/local/amber10/dat/leap/lib/all_aminoct94.lib
Loading library: /usr/local/amber10/dat/leap/lib/all_aminont94.lib
Loading library: /usr/local/amber10/dat/leap/lib/ions94.lib
Loading library: /usr/local/amber10/dat/leap/lib/solvents.lib
> source leaprc.gaff
----- Source: /usr/local/amber10/dat/leap/cmd/leaprc.gaff
----- Source of /usr/local/amber10/dat/leap/cmd/leaprc.gaff done
Log file: ./leap.log
Loading parameters: /usr/local/amber10/dat/leap/parm/gaff.dat
Reading title:
AMBER General Force Field for organic mol., add. info. at the end (June, 2003)
>
现在我们可以载入sustiva单元(sustiva.mol2):
SUS = loadmol2 sustiva.mol2
选择如果你在tleap中键入list, 应该可以看到新添加到SUS单元:
> SUS = loadmol2 sustiva.mol2
Loading Mol2 file: ./sustiva.mol2
Reading MOLECULE named SUS
> list
ACE ALA ARG ASH ASN ASP CALA CARG
CASN CASP CCYS CCYX CGLN CGLU CGLY CHCL3BOX
CHID CHIE CHIP CHIS CILE CIO CLEU CLYS
CMET CPHE CPRO CSER CTHR CTRP CTYR CVAL
CYM CYS CYX Cl- Cs+ DA DA3 DA5
DAN DC DC3 DC4 DC5 DCN DG DG3
DG5 DGN DT DT3 DT5 DTN GLH GLN
GLU GLY HID HIE HIP HIS HOH IB
ILE K+ LEU LYN LYS Li+ MEOHBOX MET
MG2 NALA NARG NASN NASP NCYS NCYX NGLN
NGLU NGLY NHE NHID NHIE NHIP NHIS NILE
NLEU NLYS NMABOX NME NMET NPHE NPRO NSER
NTHR NTRP NTYR NVAL Na+ PHE PL3 POL3BOX
PRO QSPCFWBOX RA RA3 RA5 RAN RC RC3
RC5 RCN RG RG3 RG5 RGN RU RU3
RU5 RUN Rb+ SER SPC SPCBOX SPCFWBOX SPF
SPG SUS T4E THR TIP3PBOX TIP3PFBOX TIP4PBOX TIP4PEWBOX
TP3 TP4 TP5 TPF TRP TYR VAL WAT
frcmod99SBgaff parm99
到这一步为止, 我们尚未加载parmchk2提供的frcmod文件, 因此检查我们的SUS单元就会发现缺少4个键角类型参数. 运行下面的命令进行检查
check SUS
结果如下
> check SUS
Checking 'SUS'....
Checking parameters for unit 'SUS'.
Checking for bond parameters.
Checking for angle parameters.
Could not find angle parameter: ca - c3 - c1
Could not find angle parameter: c1 - c1 - cx
Could not find angle parameter: c1 - cx - hc
Could not find angle parameter: c1 - cx - cx
Could not find angle parameter: c1 - cx - cx
There are missing parameters.
Unit is OK.
缺失的键角类型参数为ca-c3-c1, c1-c1-cx, c1-cx-hc和c1-cx-cx. 它们涉及环丙基和C-C三键的键角. 这并不意外, 因为这种体系类型在有机分子中非常少见. 现在我们可以加载frcmod文件指示tleap为缺失的键角类型使用其中的参数. 使用如下命令加载frcmod文件:
loadamberparams sustiva.frcmod
如果再次检查SUS单元, 我们应该发现已经没有缺失的参数了:
> loadamberparams sustiva.frcmod
loading parameters: ./sustiva.frcmod
Reading force field modification type file (frcmod)
Reading title:
remark goes here
> check SUS
Checking 'SUS'....
Checking parameters for unit 'SUS'.
Checking for bond parameters.
Checking for angle parameters.
Unit is OK.
现在我们就可以创建sustiva的库文件(sus.lib), 以及prmtop文件和inpcrd文件(sustiva.prmtop, sustiva.inpcrd):
saveoff SUS sus.lib
saveamberparm SUS sustiva.prmtop sustiva.inpcrd
tleap的输出中会有一些警告, 我们可以放心地忽略它们, 因为这是由sustiva中三角形键的结构导致的(只有当你理解了导致警告的确切原因时才可以忽略, 不是所有的警告都可以忽略!).
> saveoff SUS sus.lib
Building topology.
Building atom parameters.
>
> saveamberparm SUS sustiva.prmtop sustiva.inpcrd
Checking Unit.
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
1-4: angle 7 12 duplicates bond ('triangular' bond) or angle ('square' bond)
1-4: angle 7 9 duplicates bond ('triangular' bond) or angle ('square' bond)
1-4: angle 9 12 duplicates bond ('triangular' bond) or angle ('square' bond)
Building improper torsion parameters.
total 8 improper torsions applied
Building H-Bond parameters.
Not Marking per-residue atom chain types.
Marking per-residue atom chain types.
(Residues lacking connect0/connect1 -
these don't have chain types marked:
res total affected
SUS 1
)
(no restraints)
>
你可以在tleap终端中逐行输入上面的命令, 也可以将上面的命令逐行写入tleap的输入文件中(tleap.in), 然后一次生成所有需要的文件:
tleap -f tleap.in
创建Sustiva-RT复合物的拓扑和坐标文件
既然可以将传统的AMBER力场和GAFF混合使用, 我们就可以载入HIV病毒逆转录酶(RT, reverse transcriptase)的片段作为受体, 使用ff99SB力场进行处理, 同时载入Sustiva分子作为配体, 使用GAFF力场进行处理. 为此, 我们需要使用之前步骤中生成的Sustiva库文件(sus.lib).
在PDB数据库RCSB中可以找到RT-Sustiva复合物(PDB编号: 1FKO). 相应的PDB文件为1FKO.pdb.
HIV逆转录酶是异质二聚体, 由p51和p66两个亚基组成. 它是个很大的蛋白质, 分子量为117 KDa. 鉴于此教程的目的, 我们将截取与Sustiva邻近的那部分残基作为模拟体系, 包括p66亚基的指掌结构域. 截取的PDB文件为1FKO_trunc.pdb.
加载Sustiva的库文件后, 为了使tleap能够识别1FKO的PDB文件, 我们需要将1FKO PDB文件中Sustiva的残基名称从EFZ改为SUS. 由于Sustiva库文件中的原子名称与1FKO PDB文件中Sustiva分子的原子名称相同, 所以不需要进行更多修改. 在其他情况下, 始终要检查PDB文件和库文件中的原子名称是否匹配, 残基名称是否匹配. 修改后的PDB文件为1FKO_trunc_sus.pdb.
现在我们可以将PDB文件载入tleap.
首先, 如先前步骤中的那样, 我们启动tleap, 加载力场以及参数信息:
tleap -f oldff/leaprc.ff99SB
>source leaprc.gaff
>loadamberparams sustiva.frcmod
再加载Sustiva的库文件(sus.lib)和复合物的PDB文件1FKO_trunc_sus.pdb:
>loadoff sus.lib
>complex = loadpdb 1FKO_trunc_sus.pdb
最后创建截取的RT-sustiva复合物的拓扑和坐标文件:
>saveamberparm complex 1FKO_sus.prmtop 1FKO_sus.inpcrd
>savepdb complex 1FKO_sus.pdb
>quit
你可以使用VMD查看截取的RT-sustiva复合物的结构(1FKO_sus.pdb)
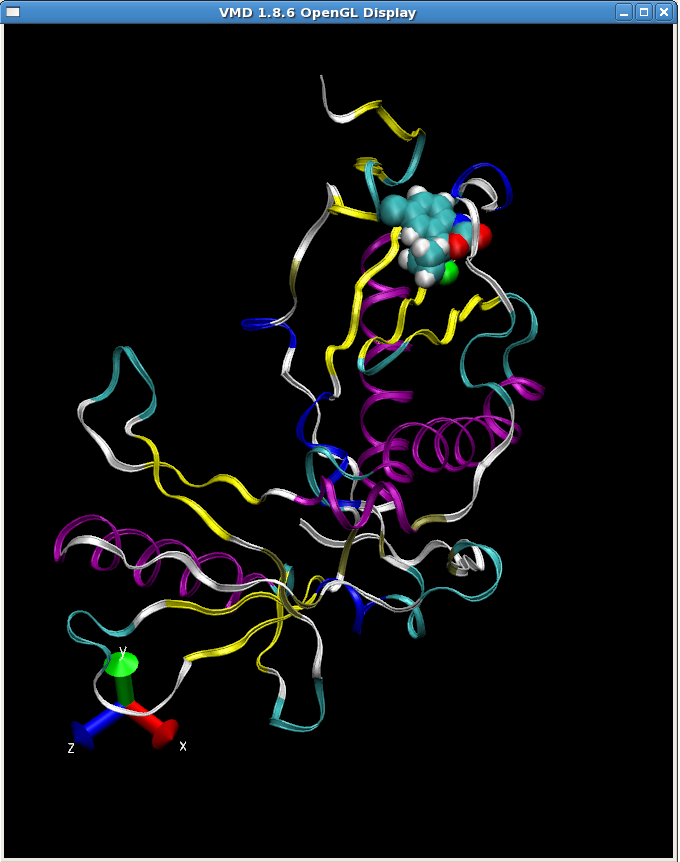
类似先前的操作, 我们将以上操作命令写入tleap输入文件(tleap2.in), 并执行生成RT-sustiva复合物的所有文件:
tleap -f tleap2.in
Sustiva-RT复合物的能量最小化和平衡
一旦有了RT-Sustiva复合物的拓扑和坐标文件, 我们就可以对其运行短时间的GB模拟了. 注意, 为了使模拟不影响完成本教程所需的总时间, 这里给出的模拟流程很短. 在真正的”成品”模拟中你应该运行足够长时间的模拟(ns)以得到好的统计收敛性.
首先, 我们将对复合物进行能量最小化以去除所有可能不合理的原子接触. 输入文件min.in如下所示:
| min.in | |
|---|---|
1 2 3 4 5 | Initial minimisation of sustiva-RT complex
&cntrl
imin=1, maxcyc=200, ncyc=50,
cut=16, ntb=0, igb=1,
&end
|
我们将进行总共200步的能量最小化(MAXCYC), 其中开始的50步使用最速下降法(NCYC), 剩下的使用共轭梯度法(MAXCYC-NCYC). 因为这不是周期性体系的模拟并且我们想要准确处理静电(NTB=0, CUT=16), 所以我们使用大的截断16埃是合理的. 对于隐式溶剂模型, 我们将使用Hawkins, Cramer和Truhlar发展的GB模型(IGB=1). 相关的完整说明见AMBER手册.
使用如下命令运行能量最小化:
sander -O -i min.in -o 1FKO_sus_min.out -p 1FKO_sus.prmtop -c 1FKO_sus.inpcrd -r 1FKO_sus_min.crd &
运行需要一定时间. 耐心等待一段时间后(在我的本地机器上花费了大约三分钟)就可得到输出文件. 如果你不愿等待, 可以下载输出文件1FKO_sus_min.out和IFKO_sus_min.crd.
如果需要, 你可以使用ambpdb生成能量最小化后的结构的PDB文件(1FKO_sus_min.pdb):
ambpdb -p 1FKO_sus.prmtop < 1FKO_sus_min.crd > 1FKO_sus_min.pdb
使用肉眼查看你的结构是一个好的习惯, 因为人类的眼睛非常善于发现模拟的异常.
下一步骤是使用热浴对RT-Sustiva复合物进行升温. 为节省教程操作时间, 我们只进行1 ps的快速升温. 实际操作中应该使用更长的时间.
我们的升温文件eq.in如下:
| eq.in | |
|---|---|
1 2 3 4 5 6 7 8 9 | Initial MD equilibration
&cntrl
imin=0, irest=0,
nstlim=1000,dt=0.001, ntc=1,
ntpr=20, ntwx=20,
cut=16, ntb=0, igb=1,
ntt=3, gamma_ln=1.0,
tempi=0.0, temp0=300.0,
&end
|
我们将运行MD(imin=0), 且不是重启运行(irest=0). 在上面的示例中我们不使用SHAKE约束, 因为氢原子的运动可能影响结合能(也可能不影响, 这里只是为了示例). 由于不使用SHAKE约束, 所以我们需要使用比常规情况更小的时间步长. 我们使用的时间步长为1 fs, 运行1000步(dt = 0.001, nstlim=1000, ntc=1, 共运行2 ps), 每20步输出一次输出文件, 每20步输出一次轨迹(mdcrd)文件(ntpr=20,ntwx=20). 我们使用Langevin动力学方法控制温度, 碰撞频率为1 ps^-1. 我们的体系从0 K开始, 目标温度为300 K(ntt=3, gamma_ln=1.0, tempi=0.0, temp0=300.0).
输入以下命令执行模拟:
sander -O -i eq.in -o 1FKO_sus_eq.out -p 1FKO_sus.prmtop -c 1FKO_sus_min.crd -r 1FKO_sus_eq.rst -x 1FKO_sus_eq.mdcrd &
升温轨迹和重启坐标分别保存在1FKO_sus_eq.mdcrd和1FKO_sus_eq.rst中. 现在sustiva-RT复合物已经进行过能量最小化和升温, 你可以查看一下体系300 K时的轨迹快照. 这个结构可以用于下一步平衡的初始结构.
参考资料
- Wang, J., Wolf, R.M., Caldwell, J.W., Kollman, P.A., Case, D.A. “Development and Testing of a General Amber Force Field”, J. Comp. Chem., 2004, 25, 1157 - 1173.
- Bayly, C.I., Cieplak, P., Cornell, W.D., Kollman, P.A. “A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges : The RESP Model”, J. Phys. Chem, 1993, 10269-10280.
- Jakalian, A., Bush, B.L., Jack, B.D., Bayly, C.I., “Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method.”, J. Comp. Chem., 2000, 21, 132-146.
译者 李睿 附言
总结
- 本教程涉及使用GAFF力场创建小分子化合物的拓扑以及相关参数文件, 小分子配体-蛋白质受体复合物参数文件的组装以及简单的amber MD模拟流程.
- 使用程序:
antechamber,parmchk2,tleap. - 处理步骤: 1. 小分子参数文件的前处理; 2. 复合物的组装; 3. 部分MD模拟.
命令及部分流程测试
注意: 我使用的是自己的体系所以名字与教程不同, 但操作流程一致.
1. 前处理
reduce part1.pdb > part.pdb
# 加H这一步非常重要, 只有使用正确加氢之后的结构进行模拟才符合实际情况.
# 每次加氢之后必须检查结构是否正确, 这是本教程的关键!
antechamber -i part.pdb -fi pdb -o pa.mol2 -fo mol2 -c bcc
# 由pdb文件生成mol2文件, -s 可以不用, 输出信息更少.
parmchk -i pa.mol2 -f mol2 -o pa.frcmod
# 检查mol2文件缺失的参数, 在frccmod文件内补全.
2. 使用tleap加载数据并进行处理和转换
tleap # 开启leap, 载入信息
source leaprc.ff99SB # 注意大小写一致
source leaprc.gaff # 加载要使用的GAFF力场
# 不理解为什么还要加载 tleap -f oldff/leaprc.ff99SB
OMET=loadmol2 pa.mol2 # 将pa.mol2文件以 OMET 名称载入leap
loadamberparams pa.frcmod # 载入缺失参数文件pa.frcmod
check OMET # 程序自检OMET分子结构
saveoff OMET OMET.lib # 将leap内OMET分子结构保存为OMET.lib文件用与复合物的组装.
saveamberparm OMET OMET.prmtop OMET.inpcrd
# 将leap内OMET分子结构保存为OMET.prmtop和OMET.inpcrd文件备用
# 两个文件扩展名也可分别为.top和.crd, 仔细看名称, 一一对应.
# 注: acpype -p OMET.prmtop -x OMET.inpcrd -d
# 将amber文件转换到gromacs文件, 这与非标准残基的处理方法相同
3. 复合物的组装
tleap # 开启leap, 载入信息
source leaprc.ff99SB # 注意大小写一致
source leaprc.gaff # 加载要使用的gaff力场.
loadamberparams pa.frcmod # 载入缺失参数补充文件
loadoff OMET.lib # 将之前处理的lib文件载入
# 注: OMET=loadmol2 OMET.mol2
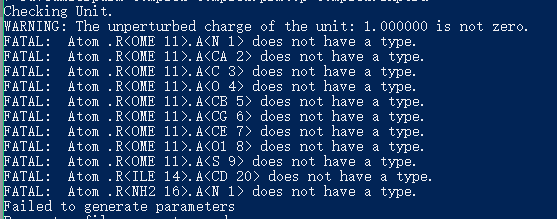
# 经测试, 如果出现如下图原子缺失错误则需要再次载入OMET.mol2, 并进行相应修改
complex = loadpdb 1FKO_trunc_sus.pdb # 将复合物模板载入leap进行识别
saveamberparm complex 1FKO_sus.prmtop 1FKO_sus.inpcrd # 输出prmtop和inpcrd文件
savepdb complex complex.pdb # 保存为pdb格式
4. 部分MD模拟
限于工作需要, 我学习amber的初衷是用amber来补充gromacs做不到的事情, (笑. 对我来说, 目前的需求在于分子模拟入门并应用, 追求准确的结果或者能被广泛接受的模拟技术流程, 而不是花式做模拟). 所以我日常工作多使用amber的免费部分进行数据处理, 转换, 以及模拟后能量分析. 长程模拟都在gromacs上进行.
总而言之, 我在使用amber进行模拟这部分基本没什么心得, 就不在此班门弄斧了.
后记
时间仓促, 翻译部分有点粗糙, 本着求真务实的态度, 请各位老师批评指正.
感谢GROMACS/AMBER中文组的各位老师同学对我学习的帮助与支持, 我相信只有互利, 才能共赢.
祝各位模拟顺利~~o( ̄︶ ̄)o.
谢谢大家!
