目标
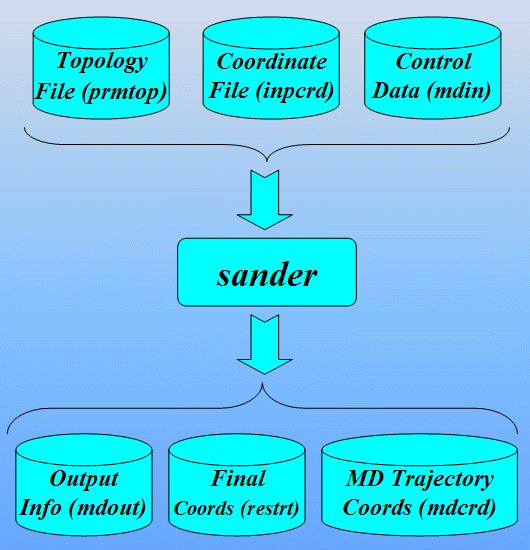
本教程这一部分介绍创建sander输入文件所需要的AMBER程序/工具, sander将使用它们执行非标准残基的能量最小化和分子动力学模拟.
本节采用的主要程序是LEAP: 它是一个读入力场, 拓扑结构和坐标信息, 并生成正式计算(即能量最小化, 分子动力学, 分析…)所需文件的程序. 该程序有两个版本, 一个是称为Xleap的图形界面和一个称为Tleap的终端界面. 既然我们要”看”模型的图形表示, 本教程将使用Xleap. 准备非标准残基参数文件的工作流程如下所示:
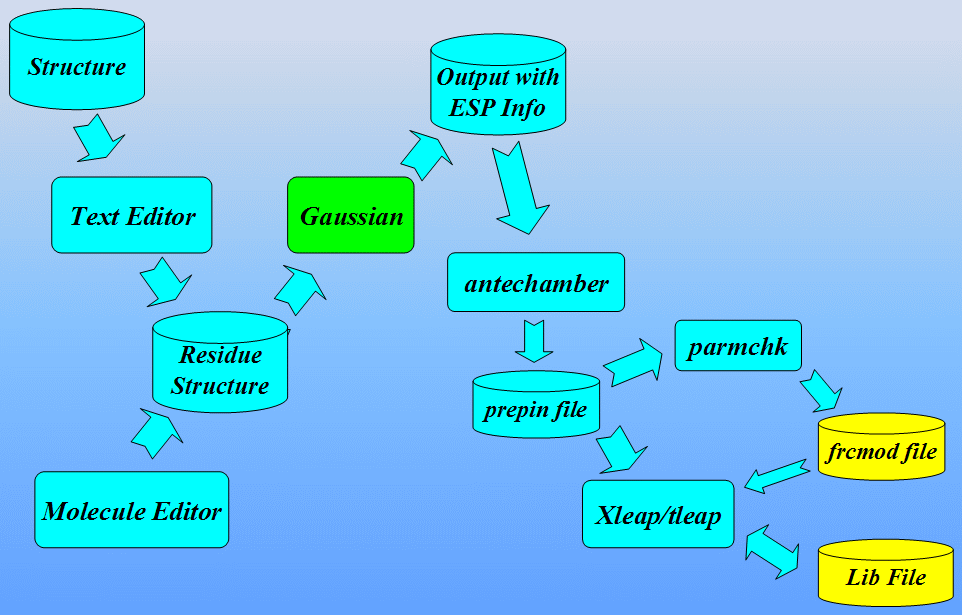
最后我们将学习如何从sander的输出文件中分析MD结果提取信息. 接着我们将使用一个叫ptraj的程序处理和分析一系列从MD轨迹输入文件中读取的3D坐标.
创建必要的拓扑和坐标文件
第一步就是为我们即将模拟的体系创建拓扑(prmtop)和坐标(inpcrd)文件. 我们将使用Xleap来完成.
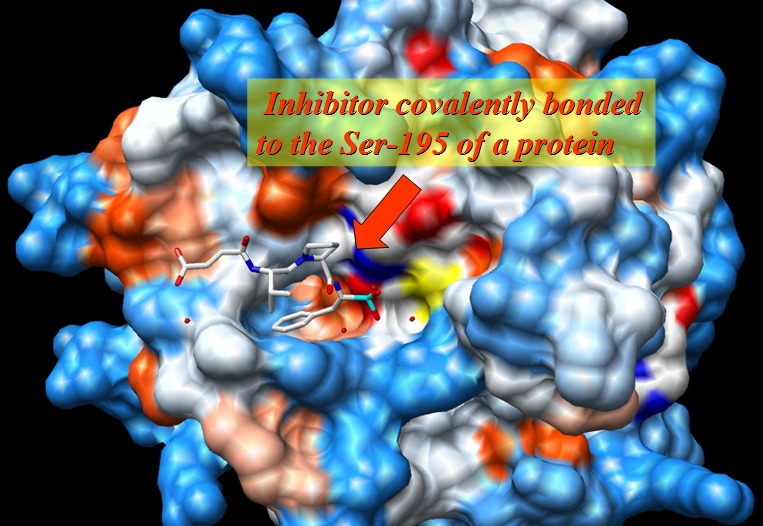
原始文件1CGH.pdb是人体组织蛋白酶G与抑制剂Suc-Val-Pro-Phep-(OPh)2形成的复合物的X射线结构, 该抑制剂与蛋白质的丝氨酸-195共价结合.
编辑PDB文件
- 阅读PDB文件的所有头部信息.
- 移除PDB尾部的所有的成键信息, 因为
Xleap不会用到它们. - 将42, 58, 136, 168, 182 和 201号半胱氨酸的残基名由CYS 改为 CYX.
- 由于原始1CGH.pdb文件中的所有组氨酸会在两处(译注: 组氨酸质子化参见教程)都发生质子化, 所以将所有的组氨酸残基名由HIS改为HIP.
- 蛋白质的丝氨酸-195残基与抑制剂的PPH残基共价结合. 可以预测磷酸化后丝氨酸-195上的电荷分布会发生很大的变化, 所以我们将其定义为一个单独的残基(本例子是SEP - 磷酸化的丝氨酸)
原始文件
ATOM 1871 N SER A 195 13.718 33.026 5.055 1.00 11.57 N
ATOM 1872 CA SER A 195 14.819 33.619 5.831 1.00 11.59 C
ATOM 1873 C SER A 195 15.463 32.581 6.740 1.00 10.79 C
ATOM 1874 O SER A 195 14.748 31.761 7.334 1.00 11.03 O
ATOM 1875 CB SER A 195 14.319 34.766 6.722 1.00 14.66 C
ATOM 1876 OG SER A 195 13.988 35.933 5.966 1.00 14.83 O
ATOM 1877 H SER A 195 12.845 33.454 5.036 1.00 20.00 H
修改后
ATOM 1871 N SEP A 195 13.718 33.026 5.055 1.00 11.57 N
ATOM 1872 CA SEP A 195 14.819 33.619 5.831 1.00 11.59 C
ATOM 1873 C SEP A 195 15.463 32.581 6.740 1.00 10.79 C
ATOM 1874 O SEP A 195 14.748 31.761 7.334 1.00 11.03 O
ATOM 1875 CB SEP A 195 14.319 34.766 6.722 1.00 14.66 C
ATOM 1876 OG SEP A 195 13.988 35.933 5.966 1.00 14.83 O
ATOM 1877 H SEP A 195 12.845 33.454 5.036 1.00 20.00 H
这里有修改后的PDB文件: 1cgh-modified.pdb.
下一步在分子编辑器中编辑修改后的PDB文件, 删除所有的氢原子和距离蛋白质3Å范围外的水分子.
这里有再次修改后的PDB文件: 1cgh-mod2.pdb.
如果此刻我们简单地将编辑后的PDB文件加载到Xleap中, 那么该文件的绝大部分都会正常加载. 但是我们非标准残基(SEP, SIN和 PPH)将会出现问题.
打开XLeap
$AMBERHOME/exe/xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff03
加载PDB文件:
x = loadpdb 1cgh-mod2.pdb
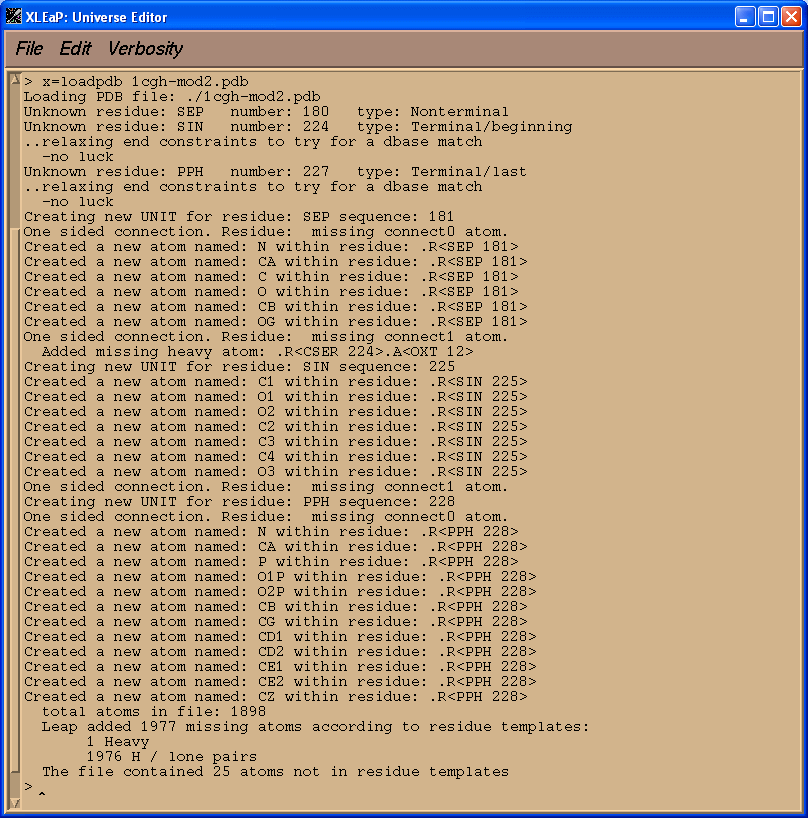
我们看到XLeap无法成功加载PDB文件. 我们需要告诉XLeap我们的非标准单元是什么, 这样才能够顺利地将修改后的PDB文件加载进去.
创建SIN单元
现在我们需要为SIN单元创建一个库文件. 因为我们需要这个单元的初始结构, 所以最简单的处理方法就是简单地把它从1cgh-mod2.pdb文件中剪切出来, 并把它保存为它自己的PDB文件.
因此, 我们将SIN和邻近的氨基酸VAL(缬氨酸)剪切出来: sin-val.pdb. 我们的目标是准备一个用高斯(Gaussian)计算RESP电荷的系统.
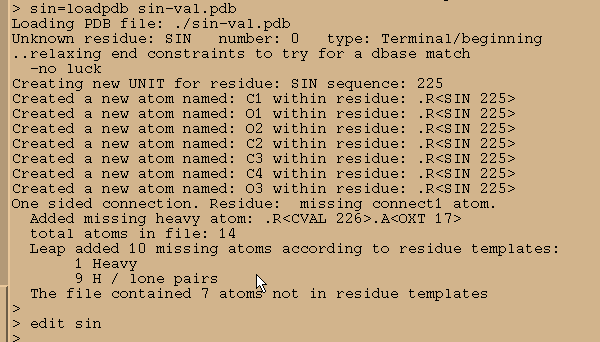
我们把它加载到Xleap并开始编辑.
sin = loadpdb sin-val.pdb
edit sin
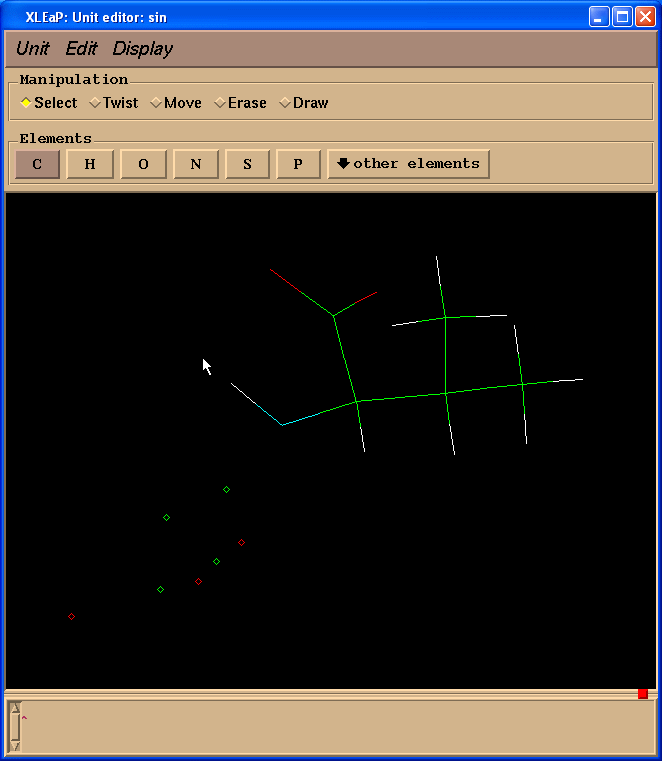
在这个窗口中, 鼠标左键允许原子选择(拖放), 鼠标中键旋转分子和鼠标右键平移分子. 如果鼠标中键不工作, 你可以通过按住Ctrl键和鼠标左键来模拟它. 为了使图像变大或变小, 同时按住中键和右键(或Ctrl和右键), 向上移动鼠标放大或或向下缩小.
如果你在使用鼠标左键选中了原子, 通过按住Shift键的同时绘制矩形选择框可以取消选择某个区域. 双击左键可以选择所有原子(双击左键的同时按住Shift键可以取消选择).
然后我们在操作(Manipulation)面板中选择绘制(Draw)复选框:
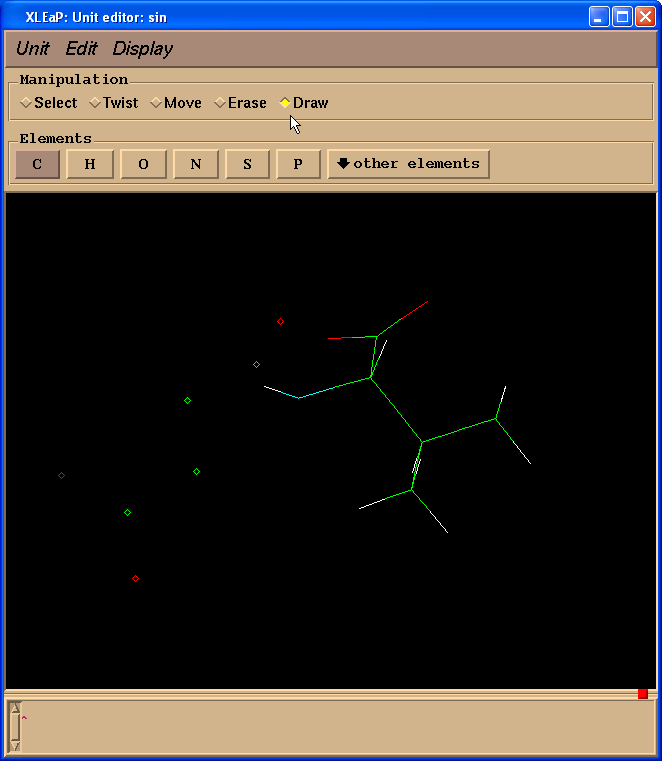
并且开始绘制缺失的键(点击原子并拖动光标到另外一个原子):
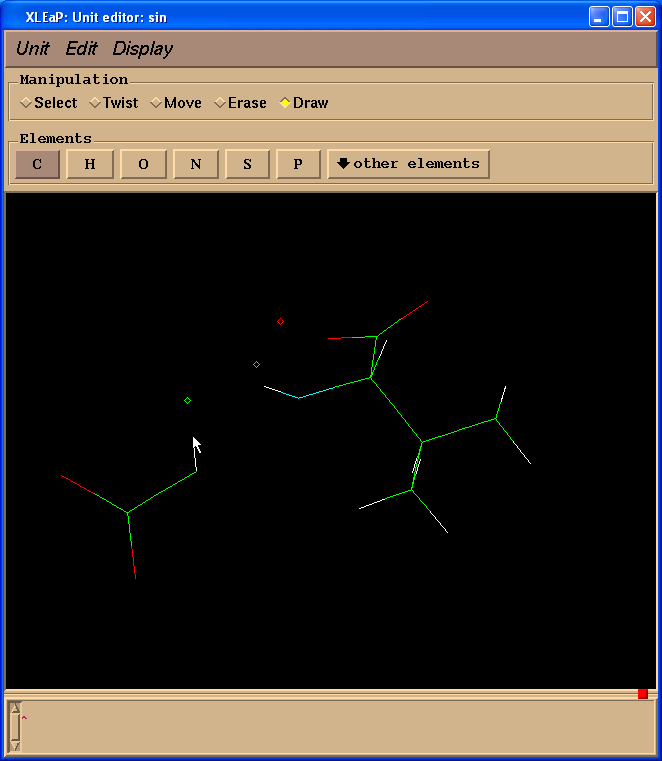
因为我们计算电荷时不需要缬氨酸残基, 而是以一个N(甲基)部分代替, 模拟一个相邻的氨基酸, 于是我们切换到擦除(Erase)模式.
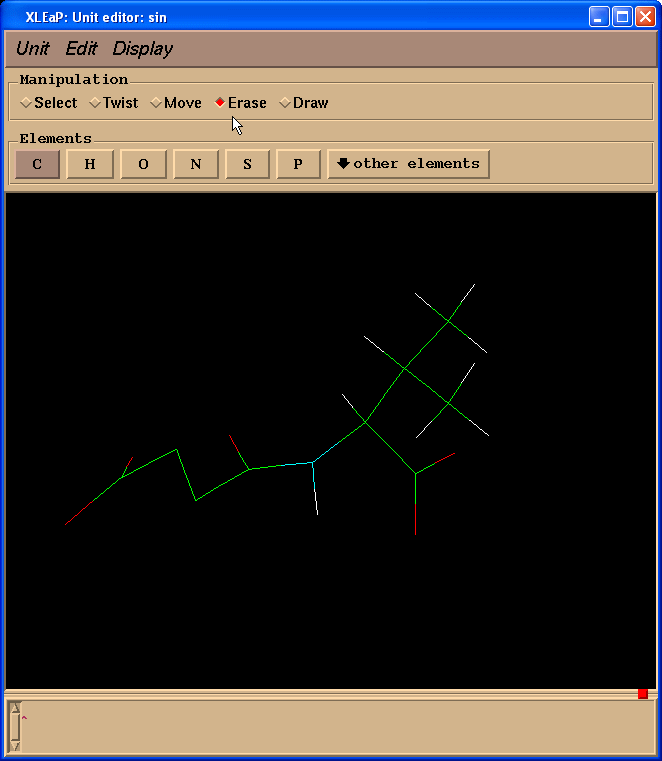
然后开始擦除缬氨酸残基的原子(把光标指向要删除的原子, 左键单击).
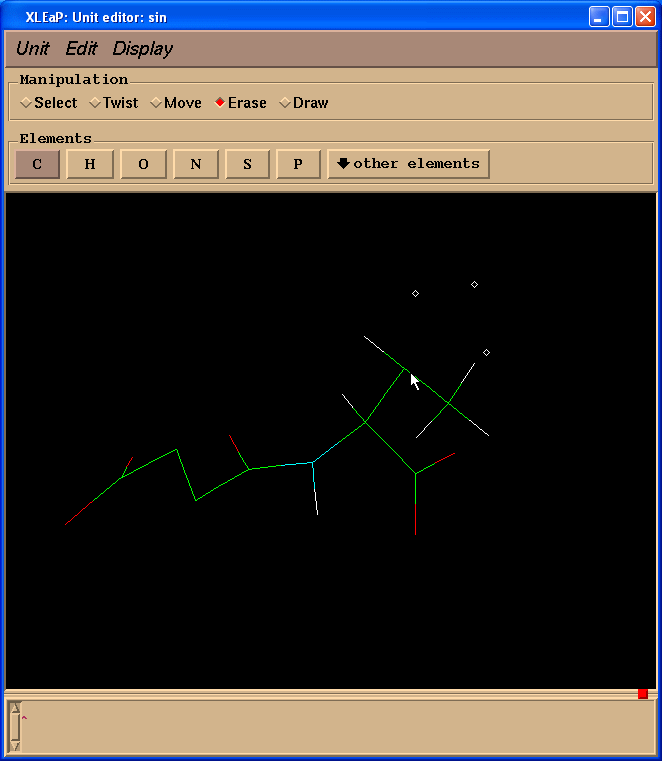
于是我们处理得到的结构就像这样:
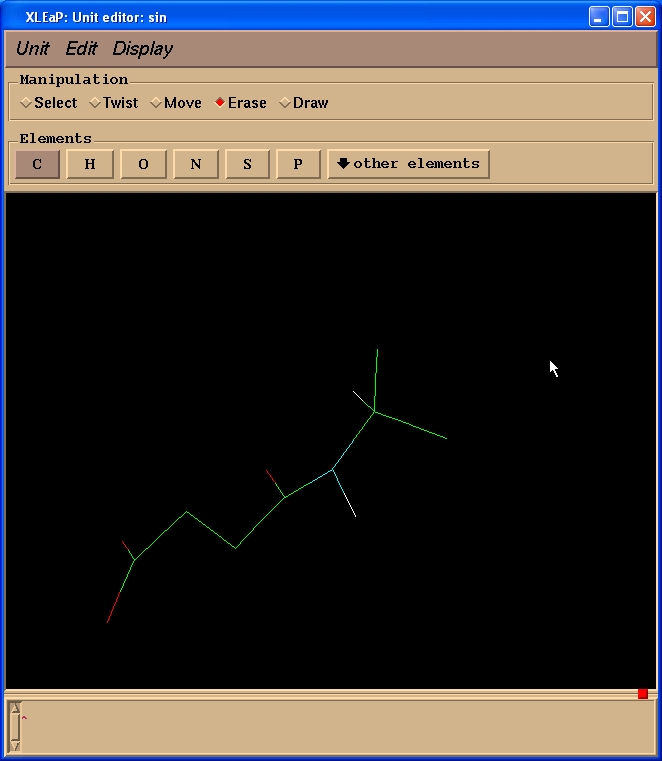
接下来我们将把两个碳原子改为成氢原子. 要做到这一点, 我们需要切换回绘图(Draw)模式, 在元素(Element)面板中选择H. 然后需要将光标指向要改变的碳原子, 左键单击:
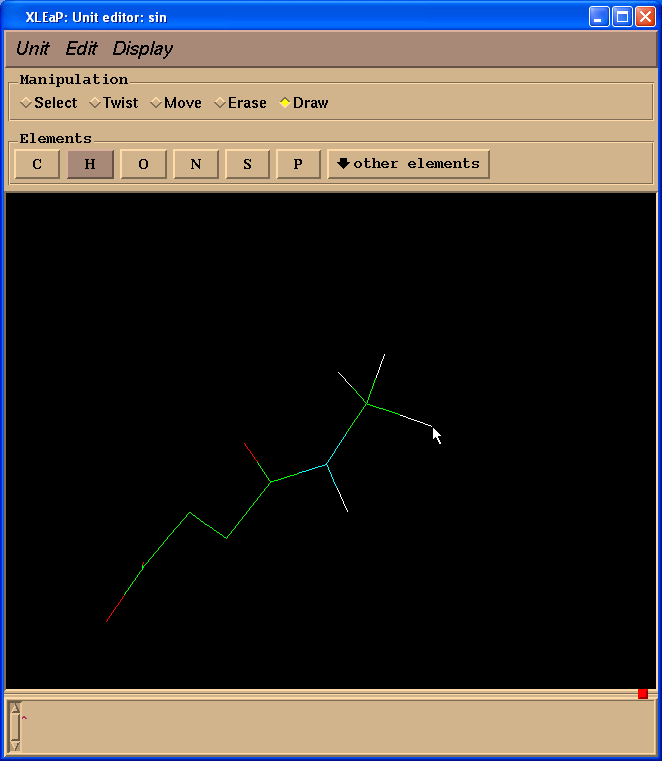
然后我们需要给SIN残基添加缺失的氢原子. 最简单的处理办法就是进入单元(Unit) -> Add H & Build菜单.
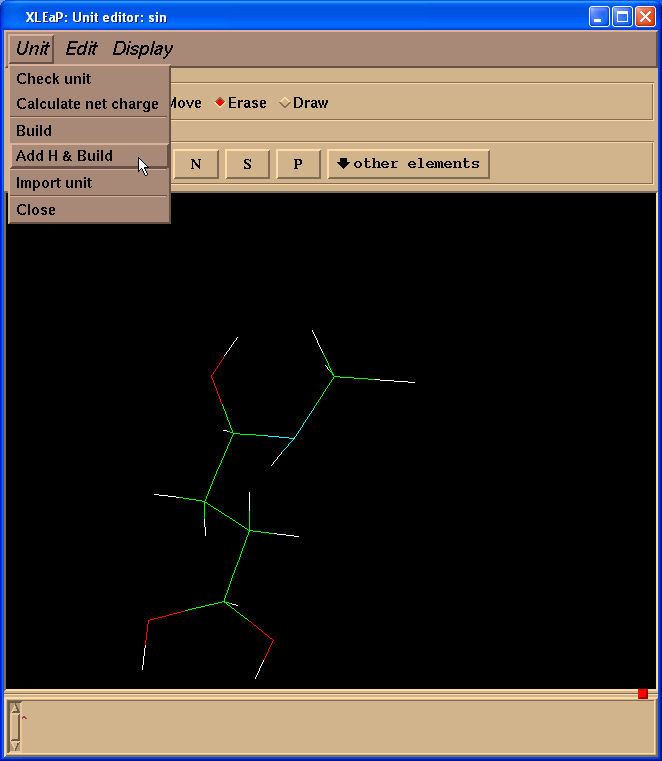
Xleap会添加超过我们需要的氢原子, 但是多余的氢原子可以很容易删除掉. 于是, 我们的最终结构就像这样:
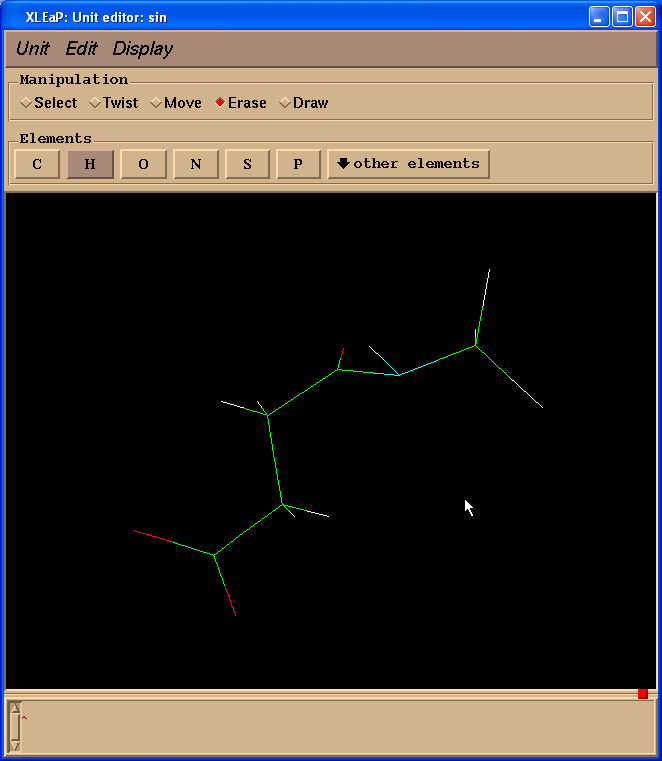
结构与理想结构相差很远, 但是我们将使用Gaussian对其进行优化.
savepdb sin sin-for-resp.pdb
我们对它进行编辑从而创建Gaussian的输入文件:
ATOM 1 C1 SIN 1 13.529 47.374 -0.405 1.00 0.00
ATOM 2 O1 SIN 1 12.629 47.356 -1.278 1.00 0.00
ATOM 3 O2 SIN 1 14.879 47.872 -0.578 1.00 0.00
ATOM 4 C2 SIN 1 13.079 46.708 0.910 1.00 0.00
ATOM 5 C3 SIN 1 14.191 45.931 1.666 1.00 0.00
ATOM 6 C4 SIN 1 13.679 45.298 2.936 1.00 0.00
ATOM 7 O3 SIN 1 13.739 45.940 3.995 1.00 0.00
ATOM 8 1H1 SIN 1 12.350 46.051 0.719 1.00 0.00
ATOM 9 2H1 SIN 1 12.740 47.407 1.539 1.00 0.00
ATOM 10 3H1 SIN 1 14.943 46.553 1.885 1.00 0.00
ATOM 11 H14 SIN 1 14.531 45.189 1.089 1.00 0.00
TER
ATOM 12 N VAL 2 12.810 44.301 2.755 1.00 0.00
ATOM 13 H VAL 2 13.554 43.738 2.367 1.00 0.00
ATOM 14 CA VAL 2 12.041 43.758 3.871 1.00 0.00
ATOM 15 HA VAL 2 12.147 44.395 4.749 1.00 0.00
ATOM 16 H18 VAL 2 12.374 42.841 4.091 1.00 0.00
ATOM 17 H19 VAL 2 11.075 43.710 3.618 1.00 0.00
TER
END
这是编辑之后的文件 (sin-for-resp.gjf):(译注: 可以用GaussView创建)
# Opt HF/6-31g* Pop=MK iop(6/33=2)
SIN
-1 1
C1 13.529 47.374 -0.405
O1 12.629 47.356 -1.278
O2 14.879 47.872 -0.578
C2 13.079 46.708 0.910
C3 14.191 45.931 1.666
C4 13.679 45.298 2.936
O3 13.739 45.940 3.995
H11 12.350 46.051 0.719
H12 12.740 47.407 1.539
H13 14.943 46.553 1.885
H14 14.531 45.189 1.089
N 12.810 44.301 2.755
H 13.554 43.738 2.367
C 12.041 43.758 3.871
H 12.147 44.395 4.749
H18 12.374 42.841 4.091
H19 11.075 43.710 3.618
Gaussian输入文件中参数的意义如下:
Opt: 我们申请对结构进行能量最小化HF/6-31g*: 我们使用6-31G*基组Pop=MK: 将会输出 Merz-Kollman 原子电荷(原子电荷符合量子化学产生的静电势)iop(6/33=2): 输出拟合静电势时的点坐标和每个点的精确静电势(译注: 参考博文: Gaussian中有用的IOp一览). 该信息在使用Antechamber生成RESP原子电荷时将会用到.
现在我们可以运行Gaussian G03程序:
g03 sin-for-resp.gjf sin-for-resp.out
我们将使用Antechamber为这个分子指定原子类型, 并计算一组RESP点电荷. Antechamber是Antechamber系列工具中最重要的程序. 它可以执行许多文件转换, 也可以分配原子电荷和原子类型. 根据输入文件的要求, Antechamber执行以下程序(AMBER 8和9均提供): divcon, atomtype, am1bcc, bondtype, espgen, respgen和prepgen. 它也会生成一系列中间文件(全部用大写字母表示).
让我们尝试对sin-for-resp.out文件中使用Antechamber. 为了创建prepin文件, 我们需要在leap中定义一个新的单元, 只需运行以下命令:
$AMBERHOME/exe/antechamber -i sin-for-resp.out -fi gout -o sin.prepin -fo prepi -c resp -s 2 -rn SIN -at amber -nc -1
这里:
-i sin-for-resp.out指定了Gaussian输出文件的名字, 包含了一个分子周围, 由量子化学计算的静电势信息.-fi gout告诉Antechamber这是一个Gaussian输出格式文件. 我们可以简单地使用其他任何支持的格式, 包括 PDB [pdb],Gaussian Z-Matrix [gzmat], Gaussian Output [gout], MDL [mdl], amber Restart [rst], Sybyl Mol2 [mol2].-o sin.prepin指定了我们输出文件的名字.-fo prepi表明我们希望输出文件为amber PREP(它是一种Leap支持的内部格式).-c resp选项告诉Antechamber计算原子点电荷时采用RESP电荷模型.-s 2选项定义了Antechamber提供的状态信息的详细程度. 本例中我们选择详细输出(2).–rn SIN定义了残基名字 SIN.–at amber定义了原子类型 (本例中为AMBER).–nc -1为分子的净电荷.
于是, 我们继续向前, 执行以上的命令. 屏幕输出应该如下所示:
Running: /opt/amber8/exe/bondtype -i ANTECHAMBER_BOND_TYPE.AC -o ANTECHAMBER_BOND_TYPE.AC -f ac -j full
Running: /opt/amber8/exe/atomtype -i ANTECHAMBER_AC.AC0 -o ANTECHAMBER_AC.AC -p amber
Running: /opt/amber8/exe/espgen -o ANTECHAMBER.ESP -i sin-for-resp.out
Running: /opt/amber8/exe/respgen -i ANTECHAMBER_RESP.AC -o ANTECHAMBER_RESP1.IN -f resp1
Running: /opt/amber8/exe/respgen -i ANTECHAMBER_RESP.AC -o ANTECHAMBER_RESP2.IN -f resp2
Running: resp -O -i ANTECHAMBER_RESP1.IN -o ANTECHAMBER_RESP1.OUT -e ANTECHAMBER.ESP -t qout
Amber8 Module: resp
resp -O -i ANTECHAMBER_RESP2.IN -o ANTECHAMBER_RESP2.OUT -e ANTECHAMBER.ESP -q qout -t QOUT
Amber8 Module: resp
Running: /opt/amber8/exe/atomtype -i ANTECHAMBER_PREP.AC0 -o ANTECHAMBER_PREP.AC -p amber
Running: /opt/amber8/exe/prepgen -i ANTECHAMBER_PREP.AC -f int -o sin.prepin -rn "SIN" -rf molecule.res
你也应该在你的目录下会得到一整系列的文件.
1cgh-mod2.pdb ANTECHAMBER_RESP1.OUT punch
1cgh-modified.pdb ANTECHAMBER_RESP2.IN qout
1CGH.pdb ANTECHAMBER_RESP2.OUT QOUT
ANTECHAMBER_AC.AC ANTECHAMBER_RESP.AC sep-and-pph-for-resp-xleap.pdb
ANTECHAMBER_AC.AC0 ATOMTYPE.INF sin-for-resp.out
ANTECHAMBER_BOND_TYPE.AC esout sin-for-resp.pdb
ANTECHAMBER.ESP leap.log sin.prepin
ANTECHAMBER_PREP.AC NEWPDB.PDB sin-val.pdb
ANTECHAMBER_PREP.AC0 pph-and-sep-in-protein.pdb
ANTECHAMBER_RESP1.IN PREP.INF
(译注: 也可以通过gesp文件直接生成mol2文件, 详细查看使用AmberTools+ACPYPE+Gaussian创建小分子GAFF力场的拓扑文件)
名字大写的的文件都是Antechamber使用的所有中间文件, 这里不再需要. 你可以安全地删除它们. 这些文件在默认情况下不会被删除, 因为如果任务不能正常完成, 这些文件可能是有用的. 我们真正感兴趣的文件是sin.prepin文件, 这也是我们当初运行Antechamber的原因. 它包含了我们SIN残基的定义, 包括所有电荷和原子类型, 我们将把它加载到Leap中并创建prmtop和inpcrd文件. 尽管在该文件中定义了最有可能的键, 角和二面角参数的组合, 但是我们的分子可能包含未被参数化的键, 角或二面角的原子类型的组合. 如果是这种情况, 那么我们必须指定所有缺失的参数, 然后才能在Leap中创建我们的prmtop和inpcrd文件.
我们可以使用parmchk功能检查所有我们需要的参数是否齐全:
$AMBERHOME/exe/parmchk -i sin.prepin -f prepi -o sin.frcmod
执行该命令后将会产生一个叫sin.frcmod的文件. 这个是一个可以被Leap加载的参数文件, 用来添加缺失的参数. 这里它将包含所有的缺失参数. Antechamber将会尽量通过类比相似的参数从而填充这些缺失的参数. 在运行模拟前你需要仔细检查这些参数. 如果Antechamber不能根据经验计算得到值或者没有类比, 它都会添加它认为合理的默认值, 要不然就插入一个占位符(所有的值都为0), 并且会加上注释”ATTN: needs revision”. 这种情况下, 你将不得不手动对其进行参数化.
我们可以看到一共缺少了9个键, 14个角和15个二面角的参数. 稍后我们将在Xleap中看看他们分别对应了哪些原子. 就本教程而言, 我们将假定Antechamber推荐给我们的参数都是可以接受的. 理论上, 你应该真正地测试这些参数(例如通过与从头算结果相比较), 从而确保它们都是合理的.
为了完成创建SIN残基, 我们将把sin.prepin加载到Xleap中, 删除多余的原子, 也就是那个相邻的”肽键”, 并把修改后的结构保存为Amber库文件.
loadamberprep sin.prepin
list
你应该可以看到一个叫SIN的新单元.
edit SIN
删除N-甲基基团:
删除N-甲基基团后, 我们应该确保我们残基的整体总电荷为-1(译注: 一般确保肽链中氨基酸单体的电荷为0, 而端残基为±1). Charge命令将会告诉我们现在的总电荷:
charge SIN

因此我们需要添加-0.0138的电荷. 为了编辑原子, 需要先选中它们(左键单击并拖动鼠标).
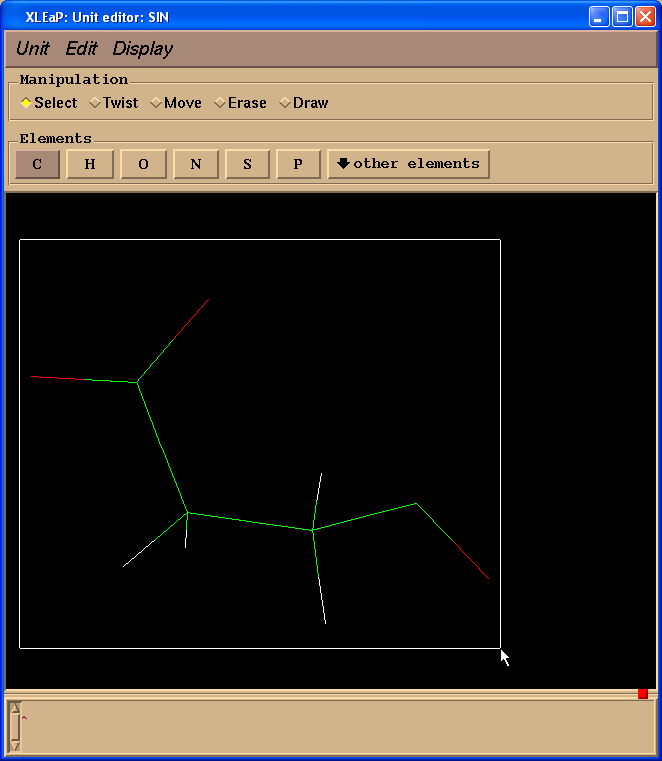
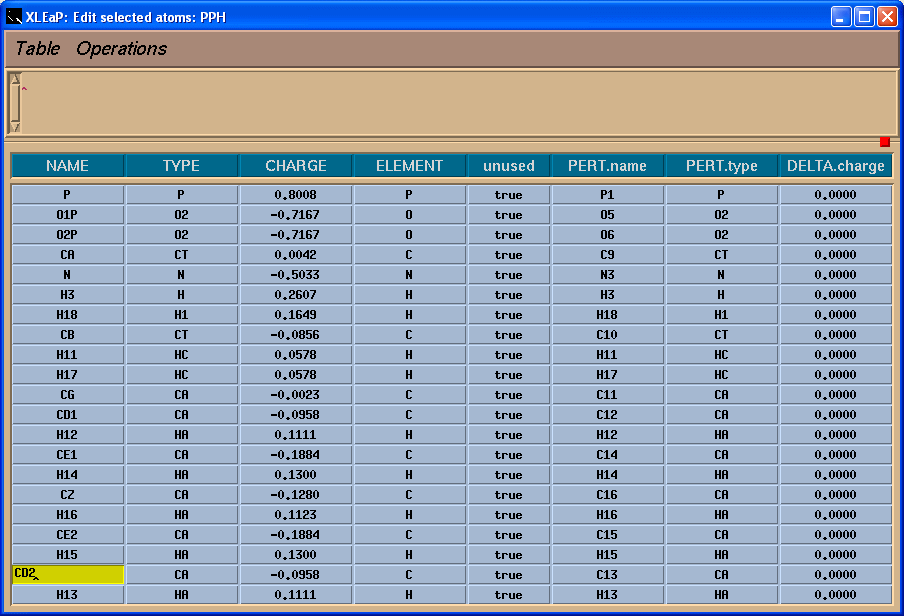
然后选择Edit -> Edit selected atoms菜单.
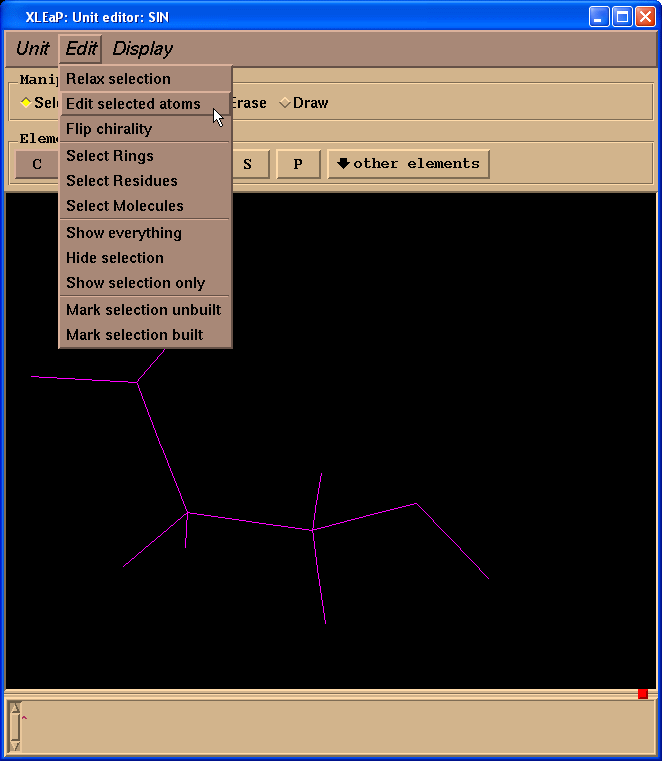
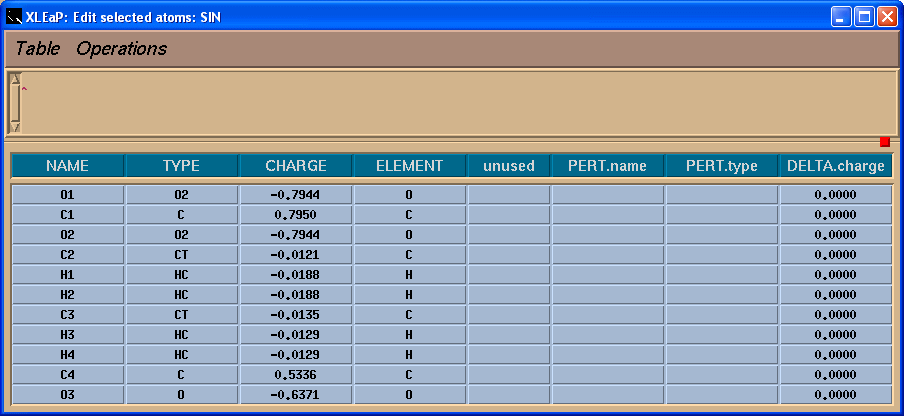
我们将看到一个表格:
左边一栏包含原子名称. 为了看到原子名称与三维结构之间的对应关系, 我们将在Xleap单元编辑器中标记原子.
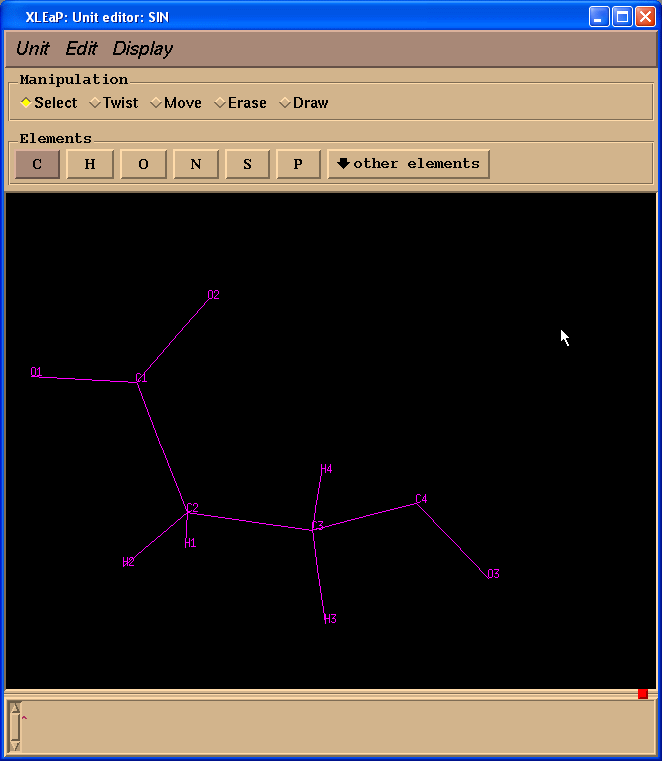
为了确保该SIN残基中的原子名称与原始的PDB文件中相同, 我们将加载预先准备的sin-val.pdb文件:
res = loadpdb sin-val.pdb
edit res
我们将看到单元编辑器的第二个窗口.
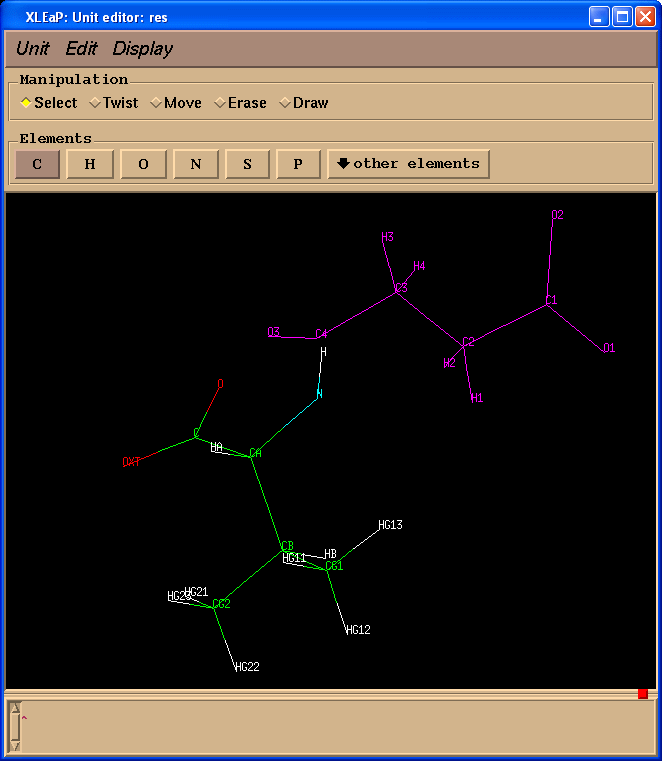
在这个特定的情况下, 两个结构中的原子具有相同的原子名称, 但这并不总是如此, 因为在准备Gaussian输入文件时, 原子的名称信息可能会丢失.
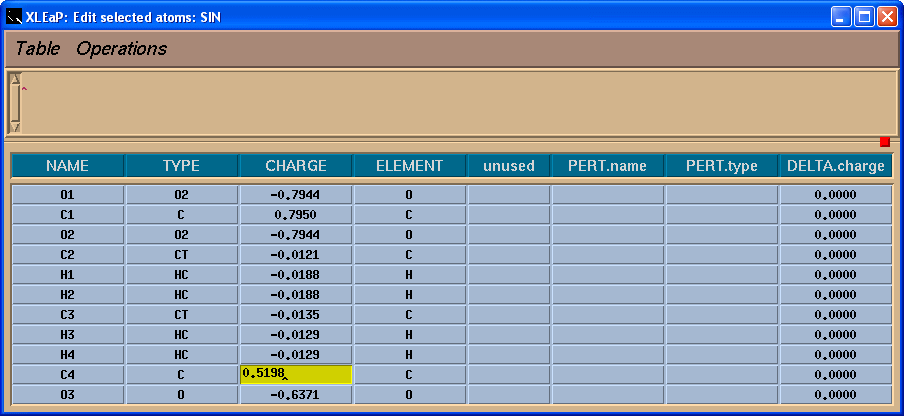
于是, 我们将把-0.0138的电荷加到C4原子上, 这样残基上的总电荷就为-1了.
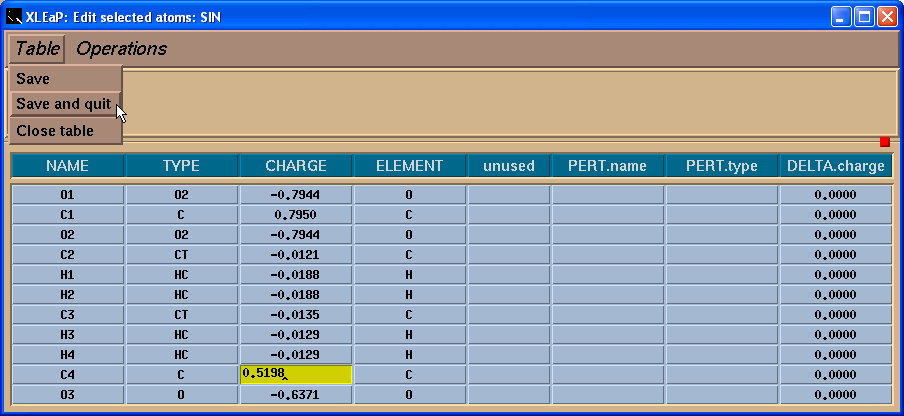
接下来我们需要保存我们更改, 点击 Table -> Save and quit 菜单:
在为已完成的单元保存库文件之前, 我们需要做的最后一件事情就是告诉Xleap这个单元的头部原子是什么以及尾部原子是什么, 这些信息用于连接蛋白质的骨架. 例如, 如果我们输入
desc SIN
我们将看到头原子没有被定义而头原子的定义不正确:

因此我们需要使用set命令为SIN单元重新指定它们. 输入:
set SIN head null
set SIN tail SIN.SIN.C4
这就是我们所做的. 现在我们可以使用saveoff命令保存已完成的库文件:
saveoff SIN sin.lib
作为最后的检查, 我们将加载原始PDB文件的一部分, sin-val.pdb, 它包含了SIN残基.
res = loadpdb sin-val.pdb
没有报出任何错误……
创建SEP 和 PPH 残基
为了创建SIP和PPH的库文件, 我们将以与创建SIN单元相同的方式开始: 简单地把它从1cgh-mod2.pdb文件中剪切出来,并把它保存为它自己的PDB文件.
于是我们剪切出SEP(之前为Ser-195), PPH和相邻的氨基酸, Val, Glu, Gly: pph-and-sep-in-protein.pdb.
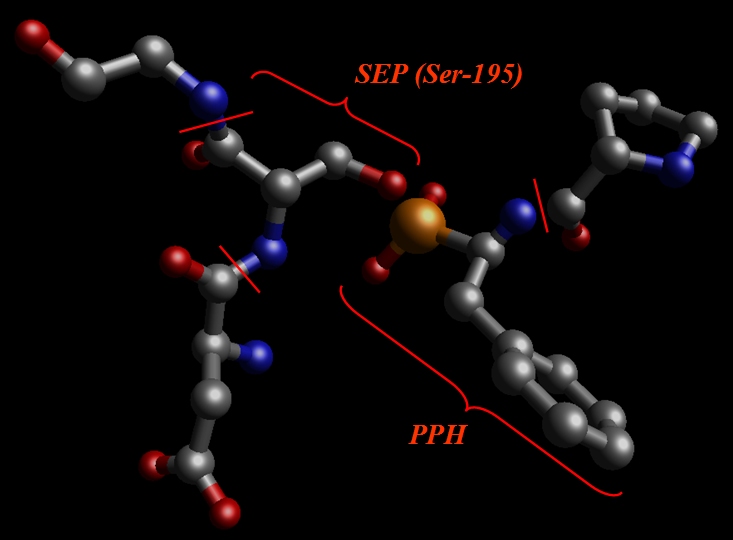
我们的目标是准备一个用高斯(Gaussian)计算RESP电荷的系统.
我们把它加载到Xleap并开始编辑.
res = loadpdb pph-and-sep-in-protein.pdb
edit res
我们将创建缺失的键(操作(Manipulation)面板中选择绘制(Draw)复选框):
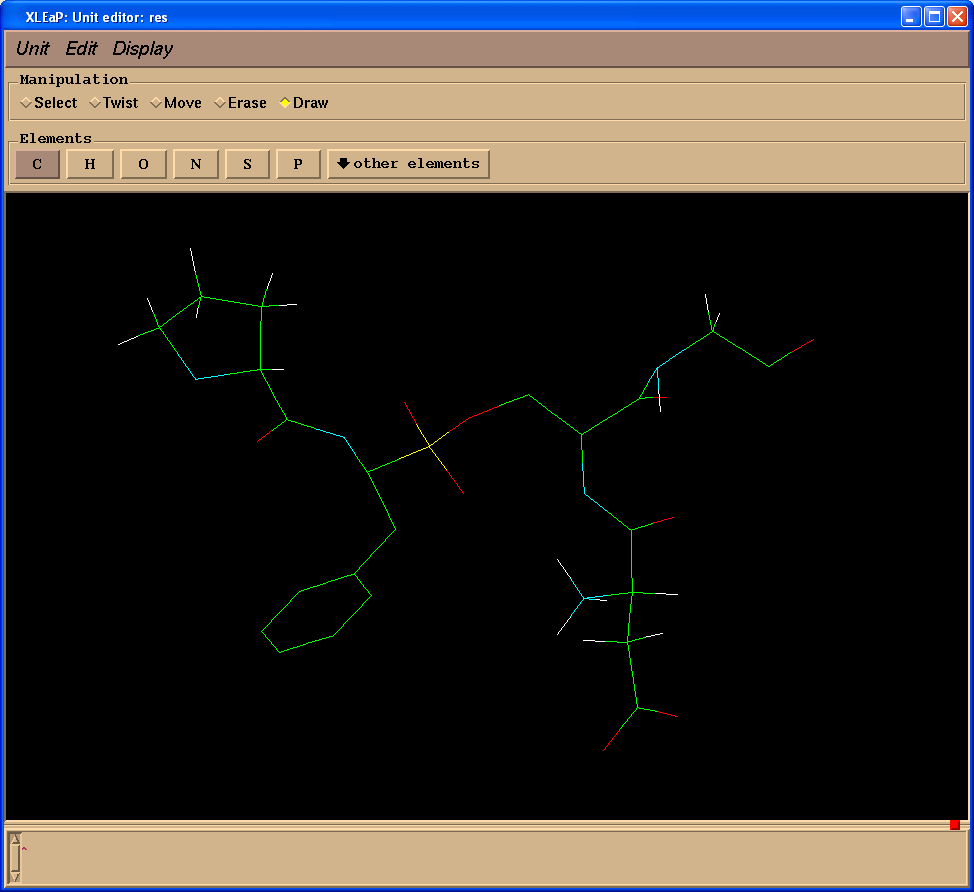
删除多余的原子(操作(Manipulation)面板中选择擦除(Erase)复选框):
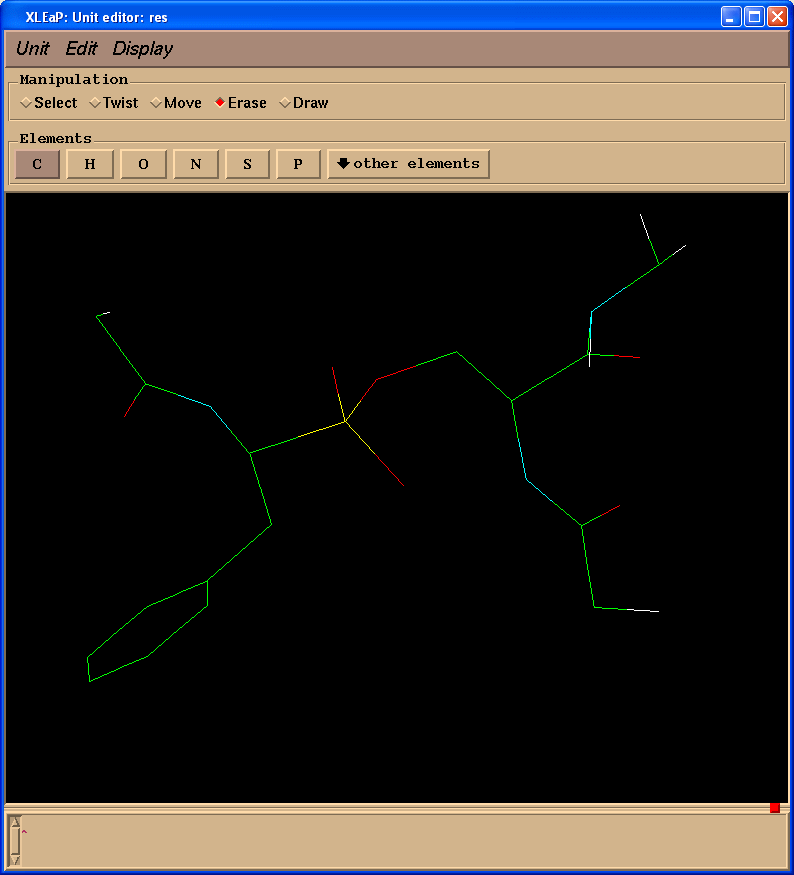
另外, 最后添加缺失的质子. 我们的最终结构看起来如下: (pph-for-resp.pdb):
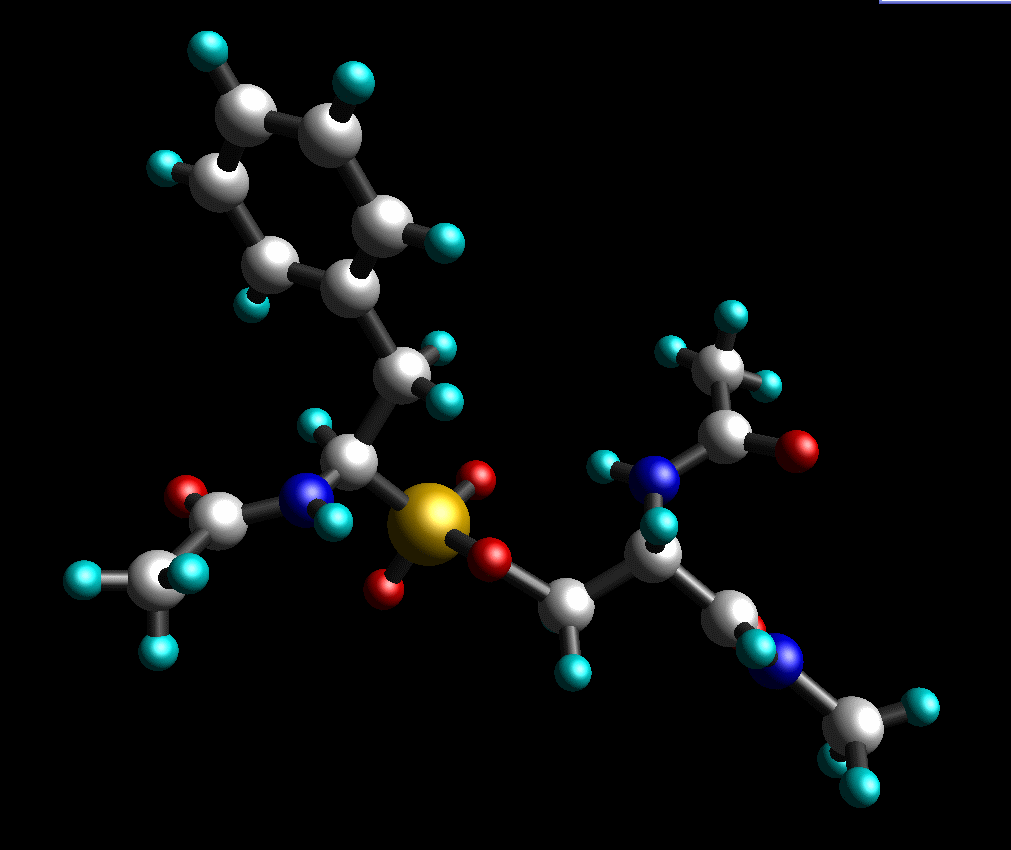
Gaussian的输入文件: pph-for-resp.gjf
g03 pph-for-resp.gjf pph-for-resp.log
运行Antechamber:
$AMBERHOME/exe/antechamber -i pph-for-resp.log -fi gout -o pph.prepin -fo prepi -c resp -s 2 -rn PPH -at amber -nc -1
它的输出:
Running: /opt/amber8/exe/bondtype -i ANTECHAMBER_BOND_TYPE.AC -o ANTECHAMBER_BOND_TYPE.AC -f ac -j full
Bond types are assigned for valence state 11 with penalty of 2
Running: /opt/amber8/exe/atomtype -i ANTECHAMBER_AC.AC0 -o ANTECHAMBER_AC.AC -p amber
Running: /opt/amber8/exe/espgen -o ANTECHAMBER.ESP -i pph-for-resp.log
Running: /opt/amber8/exe/respgen -i ANTECHAMBER_RESP.AC -o ANTECHAMBER_RESP1.IN -f resp1
Running: /opt/amber8/exe/respgen -i ANTECHAMBER_RESP.AC -o ANTECHAMBER_RESP2.IN -f resp2
Running: resp -O -i ANTECHAMBER_RESP1.IN -o ANTECHAMBER_RESP1.OUT -e ANTECHAMBER.ESP -t qout
Amber8 Module: resp
resp -O -i ANTECHAMBER_RESP2.IN -o ANTECHAMBER_RESP2.OUT -e ANTECHAMBER.ESP -q qout -t QOUT
Amber8 Module: resp
Running: /opt/amber8/exe/atomtype -i ANTECHAMBER_PREP.AC0 -o ANTECHAMBER_PREP.AC -p amber
Running: /opt/amber8/exe/prepgen -i ANTECHAMBER_PREP.AC -f int -o pph.prepin -rn "PPH" -rf molecule.res
我们可以使用parmchk功能检查所有我们需要的参数是否齐全:
$AMBERHOME/exe/parmchk -i pph.prepin -f prepi -o pph.frcmod
执行该命令后将会产生一个叫pph.frcmod 的文件. 这个是一个可以被Leap加载的参数文件, 用来添加缺失的参数.
把pph.prepin 加载到Xleap中, 删除多余的原子, 也就是那个相邻的”肽键”, 并把修改后的结构保存为Amber库文件.
loadamberprep pph.prepin
edit PPH
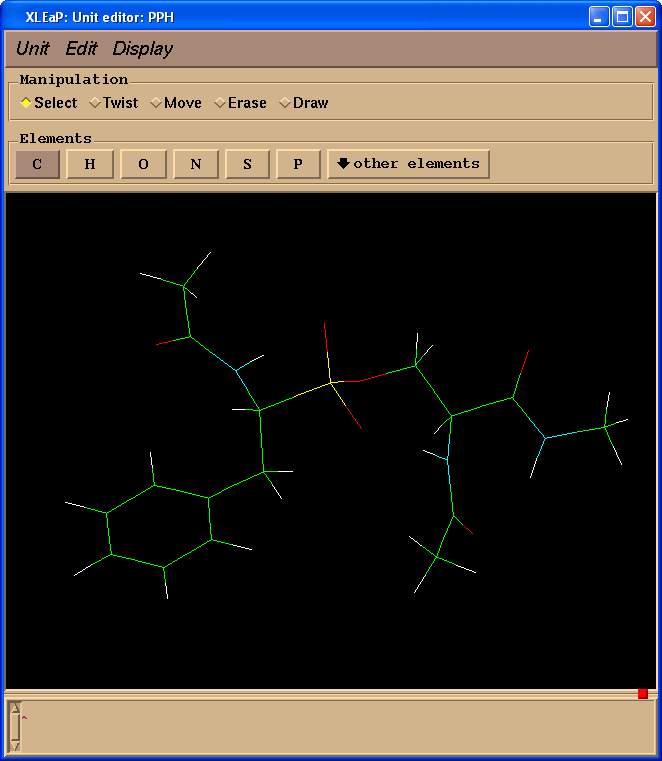
删除模拟肽键的原子(操作(Manipulation)面板中选择擦除(Erase)复选框):
统计总电荷:
charge PPH
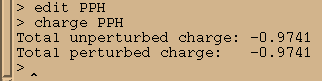
于是我们需要把-0.0259的电荷加到残基上, 我们将把它加载磷原子上:
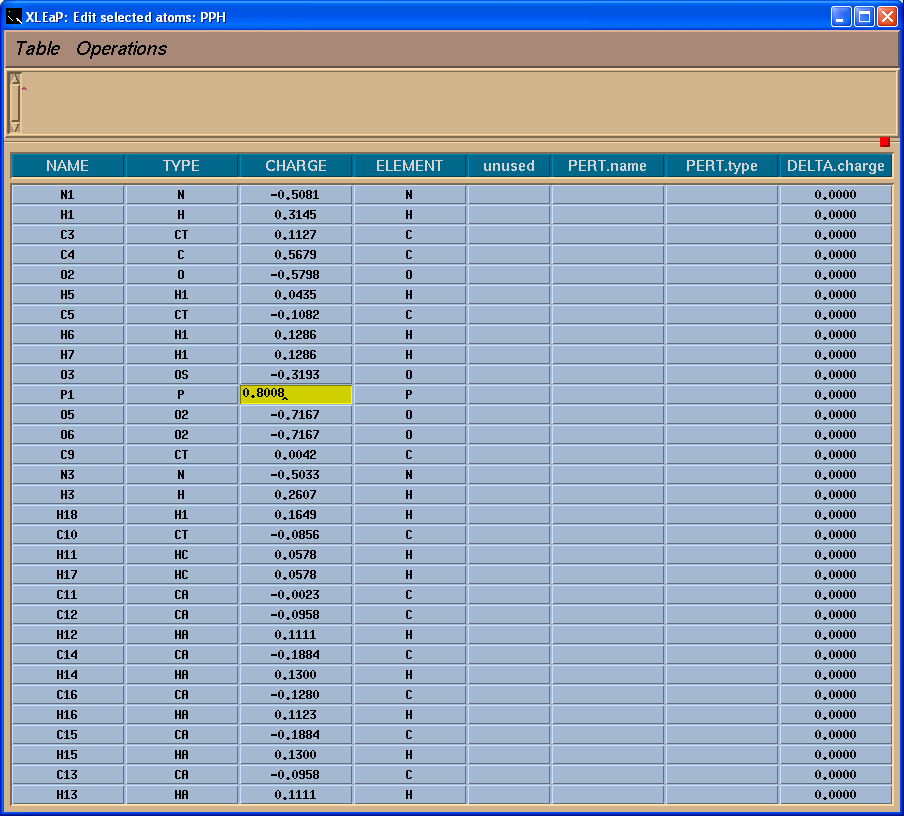
由于我们现在的单元PPH含有两个残基PPH和SEP,我们将把它分为两部分:
SEP = copy PPH
该命令创建了一个PPH的副本, 于是我们可以分开来编辑PPH和SEP残基. 开始编辑SEP并删除所有多余的原子:
edit SEP
但是存在一个问题, 我们SEP残基的原子名称与原始的pdb文件中不一致. 我们将修复它:
我们将把单元和残基命名为SEP.
sepname = SEP
set SEP name sepname
set SEP.PPH name sepname
最后一步将定义残基的头和尾原子.
set SEP head SEP.SEP.N
set SEP tail SEP.SEP.C
set SEP.SEP restype protein
最终我们将它保存为一个库文件.
saveoff SEP sep.lib
创建PPH 残基
开始编辑PPH并删除所有多余的原子.
edit PPH
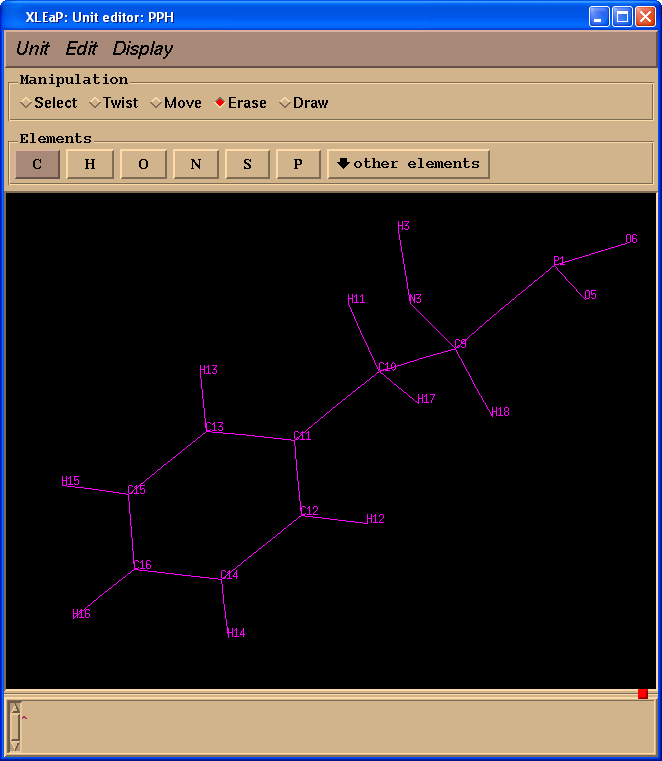
不幸的是, 在Gaussian的计算过程中, 原子名称的信息将会丢失, 所以我们需要确保我们PPH残基的原子名称与原始pdb文件中PPH残基一致. 为了更简单地完成任务, 我们将下载 pph-and-sep-in-protein.pdb.
test = loadpdb pph-and-sep-in-protein.pdb
edit test
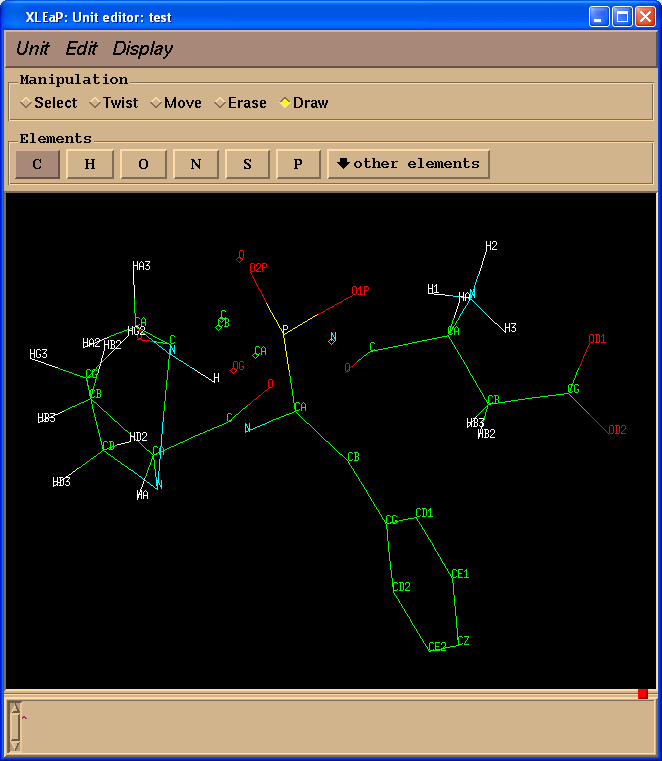
现在我么简单地重命名PPH中的原子.
最后一步我们将定义头和尾原子.
set PPH head PPH.PPH.N
set PPH tail null

最后我们将它保存为库文件.
saveoff PPH pph.lib
我们从Xleap中退出.
quit
然后重新启动.
$AMBERHOME/exe/xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff03
在我们加载PDB文件之前, 我们将加载三个残基SIN, SEP, PPH的库文件和参数文件sin.frcmod 和pph.frcmod.
loadoff sin.lib
loadoff sep.lib
loadoff pph.lib
loadamberparams pph.frcmod
loadamberparams sin.frcmod
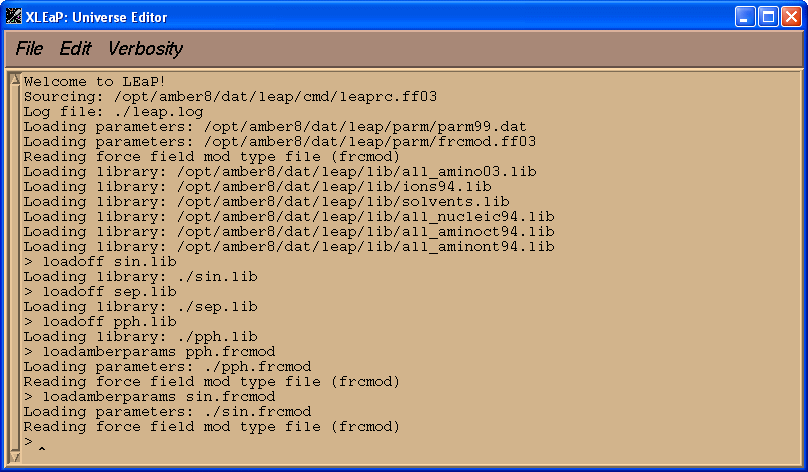
现在我们将试着加载PDB文件 1cgh-mod2.pdb.
1cgh = loadpdb 1cgh-mod2.pdb
瞧, 现在没有错误了!
最后一步我们应该使用bond命令添加Cys残基之间的三个S-S键和PPH与SEP之间的一个共价键.
bond 1cgh.29.SG 1cgh.45.SG
bond 1cgh.122.SG 1cgh.187.SG
bond 1cgh.152.SG 1cgh.166.SG
bond 1cgh.181.OG 1cgh.228.P
我们将保存蛋白质的库文件
saveoff 1cgh 1cgh.lib
为能量最小化和MD准备输入文件(隐式溶剂)
我们继续进行并在一个截断八面体的TIP3P水盒子中溶剂化我们的系统.
solvateoct 1cgh TIP3PBOX 12
我们接下来需要做的事就是添加反离子. 在我们使用”addions”命令之前, 我们需要弄清楚我们的系统是带正电还是带负电. 如果它带正电, 我们将添加负电荷的Cl-离子平衡它;如果它带负电, 我们加添加Na+离子平衡它. 为了计算我们系统的电荷, 我们可以使用”charge”或者”check”命令(见前文):
charge 1cgh
接下来我们将中和我们的系统. 因为现在它拥有+27的电荷 (check 1cgh), 因此我们一共添加27个Cl-离子.
addions 1cgh Cl- 27
保存参数:
saveamberparm 1cgh 1cgh-water-box.top 1cgh-water-box.crd
生成的拓扑文件1cgh-water-box.top和坐标文件1cgh-water-box.crd非常大.
我们可以把生成的结构保存在PDB文件, 然后之后使用现有的更复杂的可视化程序查看它.
savepdb 1cgh 1cgh-water-box.pdb
我们可以在Xleap中看看我们的系统:
edit 1cgh
在显式溶剂进行能量最小化和MD
我们将在显式溶剂中运行我们的模拟. 本章节中我们将使用prmtop和inpcrd文件( 分别为我们之前创建的1cgh-water-box.top 和1cgh-water-box.crd)
能量最小化阶段1 - 保持蛋白质固定
我们的能量最小化将包含两个阶段. 第一步中我们将保持蛋白质固定并只优化反离子和水分子的位置, 第二步中我们将优化整个系统.
在初始最小化过程中有两种保持蛋白质原子固定的方法.
一种方法是使用BELLY选项, 使用该选项后, 所有选定的原子在每一步其受力均清零. 这会导致它实质上是部分最小化. 这种方法曾经是最受欢迎的方法, 但它可能会导致不稳定性和奇怪的人为因素, 特别是在使用恒定压力周期性边界模拟的情况下, 因此, 它不再是建议的方法.
相反, 我们将在每个蛋白质原子上使用”位置限制”来使它们基本固定在相同的位置. 这种约束通过指定参考结构来工作, 在这种情况下, 我们的起始结构作为参考结构. 然后通过使用力常数限制选定的原子符合这个结构. 这可以形象地表示为利用弹簧将每个溶质原子连接到其初始位置. 从该位置移动原子会产生一个使其恢复到初始位置的力. 通过改变力常数的大小, 可以增大或减小限制作用. 这对基于同源模型的结构特别有用, 它的结构可能离平衡结构很远, 因此需要多个阶段的最小化/MD, 逐步减小力常数.
以下是我们用于溶剂和离子初步能量最小化的输入文件:
1cgh: initial minimization of solvent and ions
&cntrl
imin = 1,
maxcyc = 500,
ncyc = 250,
ntb = 1,
ntr = 1,
cut = 10
&end
Hold the protein fixed
100.0
RES 1 228
END
END
每个术语的含义如下:
IMIN = 1: 开启最小化(无MD)MAXCYC = 500: 执行总共500个步骤的最小化.NCYC = 250: 最初做250步最陡下降能量最小化, 然后是500步的共轭梯度能量最小化(MAXCYC-NCYC).NTB = 1: 使用恒定的体积周期性边界(当NTB> 0时, 在AMBER 8中PME总是“启用”的).CUT = 10: 使用10埃的截断值.NTR = 1: 根据输入文件中给出的组(GROUP)输入使用位置约束. GROUP输入在用户手册的附录中有详细描述.- 在这个例子中, 我们使用100 kcal·mol-1·angstrom-2的力常数, 并限制1到228号残基(蛋白质和抑制剂). 这意味着水和反离子可以自由移动. GROUP输入是输入文件的最后5行:
Hold the protein fixed
100.0
RES 1 228
END
END
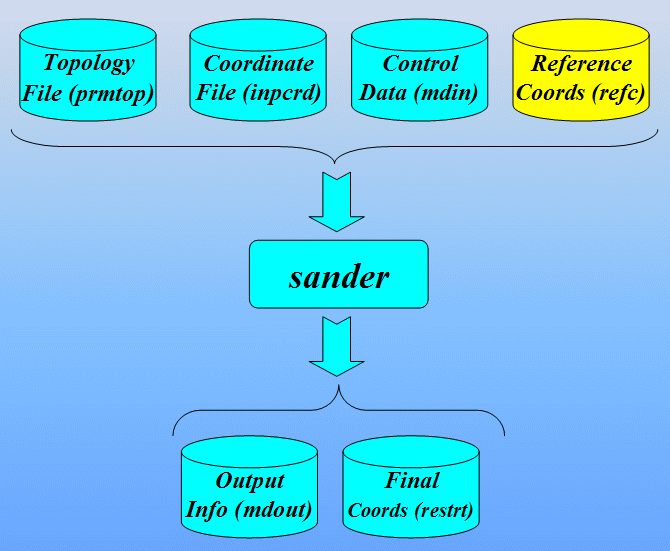
运行Sander的命令:
$AMBERHOME/exe/sander -O -i 1cgh-hold-protein.in \
-o 1cgh-hold-protein.out -c 1cgh-water-box.crd \
-p 1cgh-water-box.top -r 1cgh-hold-protein.rst \
-ref 1cgh-water-box.crd
输入文件:
输出文件:
看看这个作业的输出文件. 你应该注意起初有一个相当高的van der der Waals(VDWAALS和1-4 VDW)能量
代表水和蛋白质中的不良接触. 你还应该看到静电能量(EEL)下降得非常快. RESTRAINT能量代表我们施加的简谐限制作用. 它迅速上涨, 但后来平稳下来.
在继续之前, 为了确保它仍然合理, 我们快速浏览一下结构:
ambpdb -p 1cgh-water-box.top < 1cgh-hold-protein.rst > 1cgh-hold-protein.pdb
能量最小化阶段2 - 最小化整个系统
现在我们已经对水和离子进行了能量最小化, 下一阶段是将整个系统进行最小化. 在这种情况下, 我们将运行500步的能量最小化, 此时不施加限制. 这里是输入文件:
Entire system minimization
&cntrl
imin = 1,
maxcyc = 1000,
ncyc = 500,
ntb = 1,
ntr = 0,
cut = 10
&end
请注意, 选择要运行的能量最小化步数需要一点技巧. 运行太少会导致运行MD时出现不稳定. 运行太多将不会造成任何实际损害, 因为我们只将会越来越接近最近的局部最小点. 但是, 这会浪费CPU时间. 由于能量最小化相比于分子动力学通常要快得多, 运行比实际所需更多的最小化步骤通常是一个好主意. 在这里, 500应该够了. 我们使用以下命令来执行此操作. 请注意, 我们使用上一次运行得到的1cgh-hold-protein.rst 文件, 它包含能量最小化阶段1的最后一个结构作为最小化阶段2的输入结构. 同时, 我们也不再需要-ref选项:
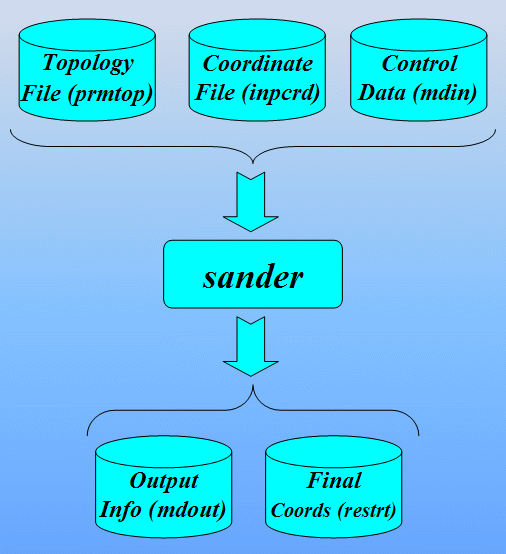
$AMBERHOME/exe/sander -O -i 1cgh-all-minimize.in \
-o 1cgh-all-minimize.out -c 1cgh-hold-protein.rst \
-p 1cgh-water-box.top -r 1cgh-all-minimize.rst
- 1cgh-all-minimize.in
- 1cgh-all-minimize.out
- 1cgh-hold-protein.rst
- 1cgh-water-box.top
- 1cgh-all-minimize.rst
分子动力学(加热)限制溶质
现在我们已经成功地将系统进行了能量最小化, 我们的平衡步骤的下一个阶段将使系统从0K升温到300K. 为了确保升温成功, 即溶质的特性不发生任何剧烈波动, 我们将使用弱约束作用, 就如我们在能量最小化阶段1中对溶质(DNA, 译注, 本教程关于分子动力学部分的阐述拷贝于Ross Walker写的TUTORIAL B1 Simulating a DNA polyA-polyT Decamer)原子所做的那样. 在AMBER 8之前, 推荐的控温方法是使用Berendsen恒温器(NTT = 1). 这种方法在很难确保系统温度保持均匀, 因此通常必须使用NMR约束, 以确保加热在20ps的时间范围内非常缓慢地进行. 这对避免产生热溶剂、冷溶质等问题至关重要. AMBER 8现在支持新的Langevin控温平衡方案(NTT = 3), 其在维持和均衡系统温度方面明显更好. 因此, 不再需要使用NMR约束. 事实上, 甚至可以简单地从300K开始升温我们的系统, 但我们将保持谨慎, 从0K开始.
注意: 你应该谨慎使用NTT = 3, 因为尽管Langevin温度调节方案可以更好地平衡温度, 但是如果作为在成品模拟中的控温方法, 它可能不太好. 使用Langevin动力学进行温度调节的经验表明, 获得准确动力学的最佳方法是使系统最初以ntt = 3达到平衡, 因为它能很好地平衡温度, 然后切换到成品时期的ntt = 1. 或者, 假设系统平衡良好, 你应该可以关闭恒温器, 只需运行显式溶剂计算, 不带恒温器(ntt = 0). 这是真实的, 因为力场是守恒的, 因此你应该能够在很长一段时间内保持恒定的能量. 然而, 积分器中的误差(事实上你做了时间步逼近)和使用截断时包含的误差最终会导致你的能量随着时间流逝而减少. 因此, 弱的控温器通常是一个好选择, 因为它纠正了这些错误.
在成品计算中使用ntt = 3的问题在于它改变了系统的动态. 实质上, ==短期动态(快速动态)==彻底发生了改变, 所以如果想获得成品阶段内系统的快速动态的信息, 那么ntt = 3就不能使用, 因为它破坏了这个信息. 而Berendsen(ntt = 1)不会这样.
对于长期动态而言, ntt = 3也会影响这些, 但是如果发生的转变不直接依赖于快速动态, 例如像A-DNA到B-DNA结构转换那样(稍后我们将在本教程中尝试), 那么ntt = 3就可以用来提高这些动态的速度. 换句话说, 你可以在更短的时间内覆盖更多的==相位空间==. 虽然langevin动力学不影响实际的转变, 但是会影响转变的速度, 因此如果你需要关于结构互转时间尺度的信息, 则需要再次使用ntt = 1. 然而, 如果你只是想快速从高能量结构转向较低能量结构, 那么ntt = 3可能是最好的方法, 但是如果你想计算诸如时间相关函数之类的东西, 那么你需要知道的是: 仔细考虑恒温器如何影响你的模拟并进行适当选择.
我们的最终目标是在恒定的温度和压力下运行成品动力学, 因为这更接近实验室的条件. 但是, 在温度较低的条件下, 也就是我们的系统处于前几ps的时间内, 压力的计算是非常不准确的, 因此在这种情况下使用恒定压力周期性边界会导致问题. 使用带有限制的恒定压力也会导致问题, 所以最初我们在恒容下运行20 ps的分子动力学. 一旦我们的系统达到了平衡(大约20 ps, 本教程为10ps), 我们将关闭约束并变为恒定压力, 然后在300 K运行另一个100 ps的平衡模拟.
由于这些模拟相比于在真空和隐式溶剂中的模拟计算成本要高得多, 所以尽量减少计算的复杂性显得至关重要. 要做到这一点, 一种方法是使用三角型的水, 即俩个氢之间的角度保持固定. 其中一个这样的模型就是TIP3P水模型, 我们用它溶剂化我们的系统. 当使用这样的水模型时, 水中的氢原子的运动也是固定的, 因为如果不这样做会导致在计算密度等方面时有非常大的不准确性. 我们的DNA氢原子的运动不太可能影响其大尺度动力学, 我们可以把这些氢都限制住. 其中一个做法是使用SHAKE算法, 即涉及到氢的键都被限制(NTC = 2). 这种去除氢运动的方法恰好可以消除系统中最高的频率振荡,也就是氢的振动. 因为它是最高频率的振荡, 它也就决定了我们时间步长的上限(在真空内运行采用1fs). 通过消除氢的运动我们可以安全地增加时间步长到2fs, 同时没有危害到我们MD模拟的稳定性. ==这允许我们可以在之前时间的一半时覆盖到给定的相空间的==,因为我们只需要50000步覆盖100ps,相反在1fs时间步时需要100,000步.
请注意, 如果您的系统非常不均匀, 您可能需要以较小的时间步长运行短时间的MD, 例如0.5fs, 然后以2fs的时间步长运行长时间的MD. 这种方法允许系统快速移除不合适的几何构型, 而不产生能量的剧烈振荡.
因此, 第1阶段运行10ps的MD, 对DNA施加弱的位置限制. 下面是输入文件:
1cgh: 1ps MD with restraints on protein
&cntrl
imin = 0,
irest = 0,
ntx = 1,
ntb = 1,
cut = 10,
ntr = 1,
ntc = 2,
ntf = 2,
tempi = 0.0,
temp0 = 300.0,
ntt = 3,
gamma_ln = 1.0,
nstlim = 5000, dt = 0.002
ntpr = 100, ntwx = 100, ntwr = 1000
/
Keep protein fixed with weak restraints
10.0
RES 1 228
END
END
每个术语的含义如下:
IMIN = 0: 关闭最小化(运行分子动力学)IREST = 0, NTX = 1: 我们生成符合Boltzman分布的随机初始速度, 只能从inpcrd(==译注: 重启文件也可以==)读取坐标. 也就是说这是我们分子动力学的第一阶段. 稍后我们将更改这些值, 以表明我们希望从离开的地方重新启动分子动力学.NTB = 1: 使用恒定的体积周期性边界(AMBER 8中当NTB> 0时, PME始终“开启”).CUT = 10: 使用10埃的截断值.NTR = 1: 根据输入文件中给出的组(GROUP)输入使用位置约束. 在这种情况下, 我们将用10埃的力常数来限制DNA.NTC = 2, NTF = 2: 开启SHAKE算法用于约束涉及氢的键.TEMPI = 0.0, TEMP0 = 300.0: 我们将温度0K开始我们的模拟, 它是由动能推导出的, 我们将使温度加热到300K. 通过调整动能, 应该可以将系统维持在300K.NTT = 3, GAMMA_LN = 1.0: 使用langevin动力学, 以1.0ps-1的碰撞频率控制温度.NSTLIM = 5000, DT = 0.002: 我们将以每步2fs的时间步长(使用SHAKE使其变得可能)运行总共5,000步的分子动力学, 总计10 ps的模拟时间.NTPR = 100, NTWX = 100, NTWR = 1000: 每100步(200 fs)写入输出文件(NTPR), 每100步写入轨迹文件(NTWX), 每1,000步写入一个重新启动文件(NTWR), 以防我们的作业发生崩溃.
我们使用以下命令运行. 注意, 我们使用我们能量最小化的第二阶段生成的restart文件, 因为这包含最后优化过的结构.
我们也把它作为限制蛋白质的参考结构:
$AMBERHOME/exe/sander -O -i 1cgh-md-restr.in \
-o 1cgh-md-restr.out -c 1cgh-all-minimize.rst \
-p 1cgh-water-box.top -r 1cgh-md-restr.rst \
-ref 1cgh-all-minimize.rst -x 1cgh-md-restr.mdcrd
- 1cgh-md-restr.in
- 1cgh-md-restr.out
- 1cgh-all-minimize.rst
- 1cgh-water-box.top
- 1cgh-md-restr.rst
- 1cgh-md-restr.mdcrd
注意: 这个模拟可能需要相当长的时间来运行(大约在一个2.66 GHz奔腾4处理器上运行2小时). 如果需要, 可以自己运行它, 或者也可以使用上面链接中提供的输出文件.
对整个系统运行平衡分子动力学
既然我们通过对蛋白质施加微弱的限制作用已经成功地在恒容下加热了我们的系统, 下一步就是改用恒压, 这样我们的水密度就可以弛豫. 同时, 现在我们已经在300K下, 可以安全地移除对蛋白质施加的限制.
我们将运行这个100ps的平衡模拟, 给予我们系统足够的弛豫时间.
1cgh: 100ps MD
&cntrl
imin = 0, irest = 1, ntx = 7,
ntb = 2, pres0 = 1.0, ntp = 1,
taup = 2.0,
cut = 10, ntr = 0,
ntc = 2, ntf = 2,
tempi = 300.0, temp0 = 300.0,
ntt = 3, gamma_ln = 1.0,
nstlim = 50000, dt = 0.002,
ntpr = 100, ntwx = 500, ntwr = 1000
/
每个术语的含义如下:
IMIN = 0: 关闭最小化(运行分子动力学)IREST = 0, NTX = 7: 我们想重新启动我们的MD模拟, 我们之前是在10ps的恒容模拟之后离开的. IREST 告诉Sander要重新启动模拟, 所以时间不重置为零, 而是从10 ps开始. 以前我们将NTX设定为默认的1, 这意味着只从restart文件中读取坐标. 然而这次, 我们想要从已完成的地方继续前进, 所以我们设定NTX = 7. 这意味着坐标, 速度和盒子信息将从一个格式化的(ASCII)重启文件中读入.NTB = 2, PRES0 = 1.0, NTP = 1, TAUP = 2.0: 使用平均压力为1atm的恒定压力周期边界(PRES0)和各向同性 的位置缩放来维持压力(NTP = 1), 并应使用2ps的弛豫时间(TAUP = 2.0)).CUT = 10: 使用10埃的截断值.NTR = 0: 我们不再使用位置限制.NTC = 2, NTF = 2: 开启SHAKE算法用于约束涉及氢的键.TEMPI = 300.0, TEMP0 = 300.0: 在MD的第一阶段中我们的系统已经加热到了300K. 所以这里就将从300K开始并维持在300K.NTT = 3, GAMMA_LN = 1.0: 使用langevin动力学, 以1.0ps-1的碰撞频率控制温度.NSTLIM = 50000, DT = 0.002: 我们将以每步2fs的时间步长(使用SHAKE使其变得可能)运行总共5,0000步的分子动力学, 总计100 ps的模拟时间.NTPR = 100, NTWX = 500, NTWR = 1000: 每100步(200 fs)写入输出文件(NTPR), 每500步写入轨迹文件(NTWX), 每1,000步写入一个重新启动文件(NTWR), 以防我们的作业发生崩溃.
我们使用以下命令运行. 注意, 我们使用我们MD的第一阶段生成的restart文件, 因为这包含最后的MD的结构, 速度和盒子信息.
$AMBERHOME/exe/sander -O -i 1cgh-equilib-md.in \
-o 1cgh-equilib-md.out -c 1cgh-md-restr.rst \
-p 1cgh-water-box.top -r 1cgh-equilib-md.rst \
-x 1cgh-equilib-md.mdcrd
- 1cgh-equilib-md.in
- 1cgh-equilib-md.out
- 1cgh-md-restr.rst
- 1cgh-water-box.top
- 1cgh-equilib-md.rst
- 1cgh-equilib-md.mdcrd
注意: 这个模拟可能需要相当长的时间来运行(大约在一个2.66 GHz奔腾4处理器上运行23小时). 如果需要, 可以自己运行它, 或者也可以使用上面链接中提供的输出文件.
分析测试平衡的结果
既然我们已经平衡了我们的系统, 那么在继续运行任何成品MD模拟之前, 检查是否已经成功达到了平衡就至关重要, 在成品模拟中我们希望了解有关我们分子的新信息.
我们应该监测一些系统属性以检查该平衡的质量. 这些包括:
- 势能, 动能和总能量
- 温度
- 压力
- 体积
- 密度
- RMSD
以下步骤将说明如何提取这些数据并对其进行绘图, 并讨论我们期望看到的成功的平衡.
分析输出文件
在本节中, 我们将看看100ps的MD模拟的系统属性, 这些可以从写入mdout文件的数据中提取出来. 这包括势能, 动能和总能量, 温度, 压力, 体积和密度. 这里是文件1cgh-equilib-md.dat, 其中分别包含时间, 温度, 压力, 总能量, 势能, 体积和密度.
让我们绘制其中的一些数据来看看我们是否已经成功达到了平衡. 首先是能量, 势能和总能量:
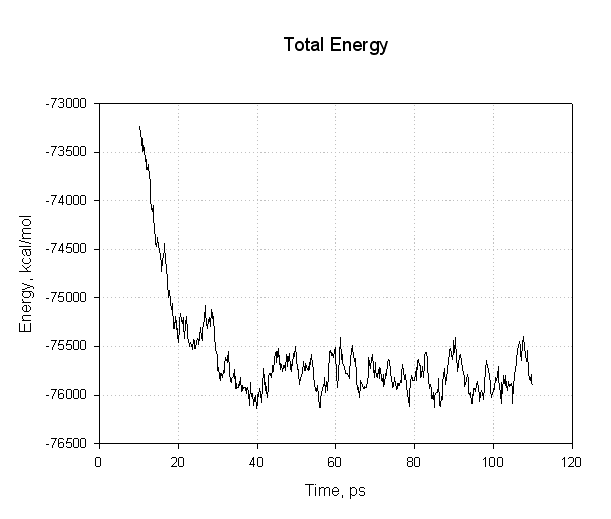
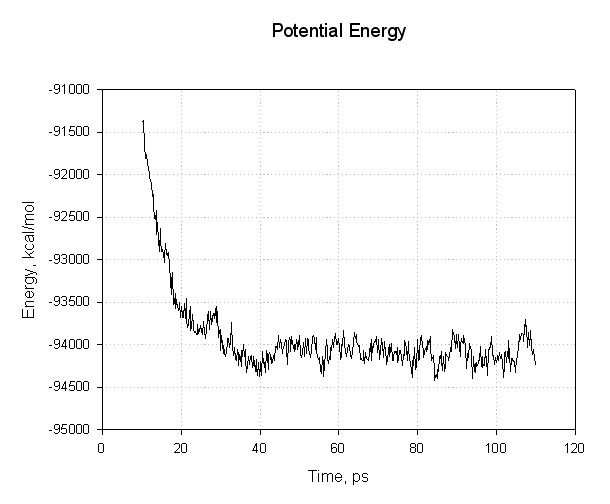
这个图表需要注意的重点是, 所有的能量在前30ps内下降. 势能和总能量开始下降, 然后在模拟的其余部分(40至110 ps)处于平稳状态, 表明弛豫已完成, 我们达到了平衡.
接下来我们绘制温度:
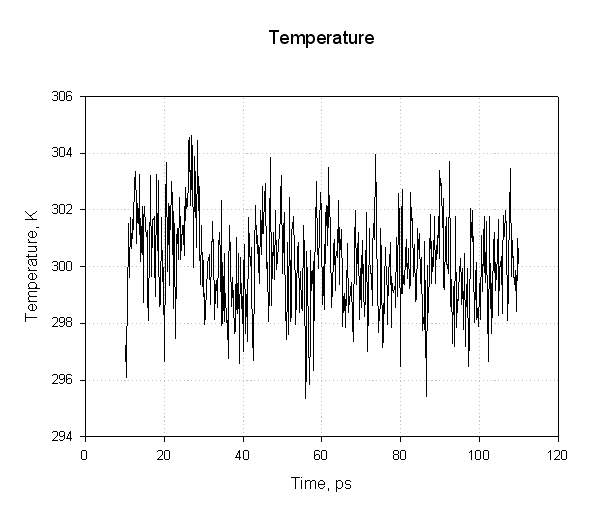
我们发现在模拟过程中温度保持不变, 说明使用langevin动力学进行温度调节是成功的.
接下来是压力:
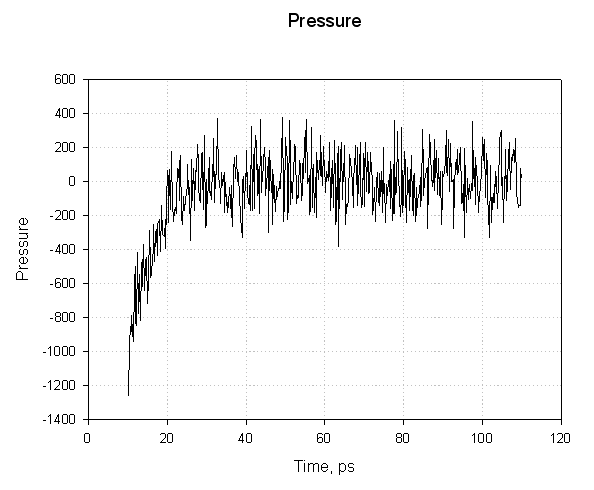
对于前20ps, 压力为负值, 并急剧增加为正值. 负压力对应于用于减小盒子尺寸的“力”而正压力对应于用于增加盒子尺寸的“力”. 这里的重点是, 虽然压力图似乎表明在模拟过程中压力波动剧烈, 但在大约20ps的模拟后, 平均压力稳定在1个大气压左右. 这足以表明它是成功的平衡.
接下来我们绘制体积:
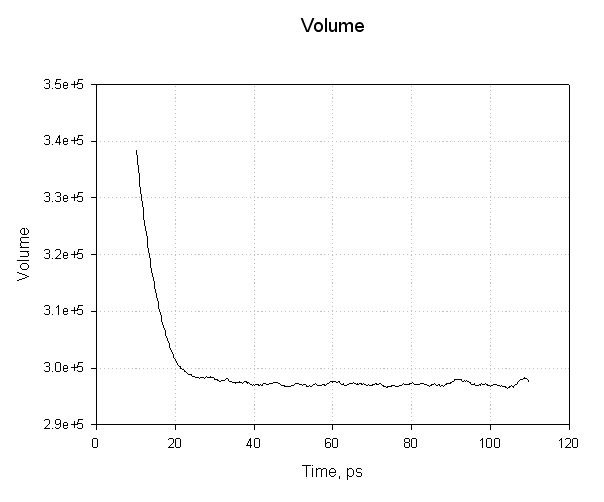
注意我们的系统体积(以埃为单位)最初是如何减小的, 我们的水盒子弛豫并达到平衡密度, 从而达到平衡体积. 这个图中的平滑过渡以及平均值附近的摆动表明我们的平衡已经成功.
最后, 我们来看看密度, 我们预计这会与体积变化相反.
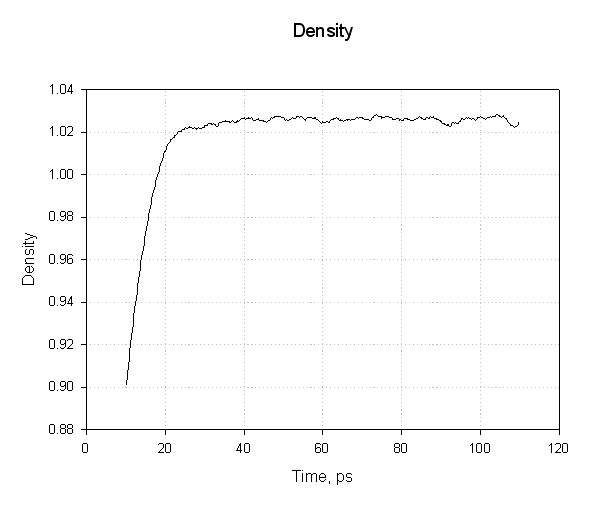
我们的系统在约1.025 g·cm-3的密度下达到平衡. 这似乎是合理的. 300K下纯液态水的密度约为1.00g·cm-3, 因此添加蛋白质和相关电荷会使密度增加约2.5%.
分析轨迹
现在我们已经分析了我们的输出文件, 并看到主要系统属性表明我们的平衡已经成功, 下一步就是考虑结构. 他们保持合理吗?需要考虑的一个有用措施是距离起始结构的均方根偏差(RMSd). 我们可以使用ptraj为我们计算这个作为时间的函数. 在这种情况下, 我们对蛋白质的骨架感兴趣, 所以我们只考虑主骨架原子C, CA和N.我们将以最终最小化后的结构为参考进行标准(而非质量加权)RMS拟合. 使用文件1cgh-all-minimize.rst作为参考). 请注意, 这次时间设置为1, 因为虽然我们每500步写入轨迹文件, 但时间步长为2 fs-因此(轨迹文件的)100步 = 1 ps. 这是ptraj的输入文件:
trajin 1cgh-equilib-md.mdcrd.gz
reference 1cgh-all-minimize.rst
rms reference out 1cgh-backbone.rms @C,N,CA time 1
让我们运行ptraj计算蛋白质骨架原子的RMSd:
$AMBERHOME/exe/ptraj 1cgh-water-box.top < analize-md.in
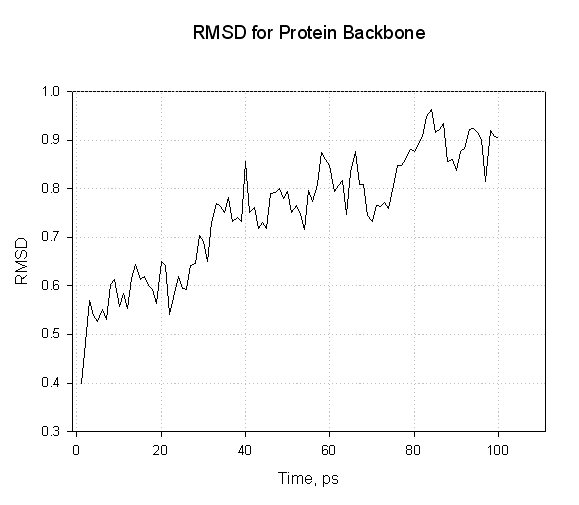
在这里, 我们看到蛋白质骨架原子的RMSd随着蛋白质在溶剂内松弛而缓慢增加. 我们没有看到任何剧烈的振荡.
