- 2018-06-07 22:38:47 翻译: 吴伟
所用程序: AmberTools 17 Linux 版
我从GLYCAM网站上找到了[给多糖残基添加化学衍生物的方法 (http://glycam.org/docs/help/2014/04/04/adding-chemical-derivatives-to-glycam-residues/), 大概翻译如下.
给多糖残基添加化学衍生物
此教程适用于GLYCAM06-h及之后的残基库中的残基, 还要求您使用GLYCAM prep文件数据库中已经存在的残基. 本教程中还有一些修改已经存在的残基的简要说明. 注: 如果残基不存在, 则需要自己创建基于GAFF力场的衍生物残基, 具体做法见后文.
向多糖残基添加衍生物的方法和建立支链结构很相似, 稍复杂的部分是当衍生物加入之后, 要对残基的电荷分配做适当的调整. 下表列出了可用的衍生物和其它的一些有用信息. 请查看以下教程, 明白怎么样去使用表中的这些信息.
| 衍生物 | 编码 | 连接原子1 | 调整电荷的部分 | 电荷调整量 |
|---|---|---|---|---|
| 乙酰化 | ACX | O | 糖环上连接的碳2 | +0.008 |
| 甲基化 | MEX | O | 糖环上连接的碳2 | -0.039 |
| 硫酸化 | SO3 | O | 连接的氧 | +0.031 |
| N | 连接的氮 | 变化的3 | ||
|
1 "O"指的是糖基上的羟基氧, "N"指的是环外的sp2的氮 2 连接在连接原子上的碳原子, 举例来说, 比如你将残基连接在三号位上的氧, 那么调整糖环三号位上的碳原子的电荷. 3 见下面的解释 | ||||
请先确认你已经有prep文件和参数
- 以上列出的衍生物已经存在于以06h开头的GLYCAM prep文件中
- 注意, 06h开头的文件中, 很多原子类型的名称已经改变, 因此, 06h和以后的文件不再与之前的版本兼容
- 请访问<glycam.org/params>获得最新的版本
下文概述了我们确定最佳电荷调整的程序
大多数多糖残基的设计是十分模块化的, 当然也是很符合实际的. 为了迎合这个目的, 当在氧原子上连接其它衍生物残基时, 需要调整连接原子的电荷, 在与氧原子相邻的碳原子上+0.194, 在氧原子上-0.194, 这样确保残基的有效性. 然而, 本教程中的衍生物与糖苷键有着本质的不同, 我们无法保留实际的电荷和模块化的”0.194”. 为了不完全放弃模块化, 我们对不同衍生物确定了简单的、可选择性的电荷变化方法. 程序选择了构建仍然是简单的、模拟结果仍然是符合实际的方法. 为了确定调整电荷的最佳过程, 我们研究了从量子力学计算到合适的模型化合物集合的电荷分布. 这就是我们如何确定的主要思路, 举例来说, 当添加一个甲基衍生物时, 最好是改变相邻碳原子的电荷而不是与之直接相连的氧原子.
计算表中电荷调整的说明:
-
这些电荷计算说明是给已经配置好的需要适当的支化结合的多糖残基准备的. 可以看下面关于选择合适的残基的O-硫酸化的例子. 当多糖残基不存在时, 可以使用一个呋喃醛糖在两个位置配以支链和/或衍生物, 其中一个是5号位.
-
如果你想对残基库中没有的多糖残基进行衍生物位置的支化, 你仍然可以使用这些计算说明, 但是你必须先调整羟基氧上的电荷, 使得模块化仍然被保持, 而且需要删掉氢原子, 脱氢应该使用tleap或者xleap程序完成, 而不是直接从输入文件中直接删除. 氧的调整电荷(
adjOxCh)是:adjOxCh = oldOxCh + HydCh – 0.194, 其中,oldOxCh是氧原子未调整时的电荷,HydCh是将要删除的羟基氢的电荷. 下面的说明描述了怎样确定adjOxCh做的进一步调整:- 计算衍生物残基的总电荷(rch), 这仅仅是衍生物残基的总电荷, 比如, 对于硫酸化残基, 在本文撰写时, 总电荷是-0.837
- 确定衍生物期望的正式电荷(fch). 这个数字应该是一个整数, 比如, 添加一个SO3-基团带来一个单位的负电荷, 添加一个NH3+带来一个单位的正电荷. 而大多数的衍生物, 比如甲基, fch=0.
- 找出rch和fch之间的差值dch, 比如说对于硫酸根, fch应该是-1.000, dch=fch-rch=-0.163, 也就是说, 多糖分子应该抵消了硫酸根电荷的大部分.
- 找出多糖残基在连接点的固有电荷ich, 这一步有点不同, 取决于你连接的类型
- 当你将衍生物连接到羟基氧上时, ich的值就是-0.194, 因为上文提到的模块化的问题. 备注: 你必须使用用于连接单糖的GLYCAM残基, 正如下面的硫酸化的教程一样, 也就是说, 比如你想在α-D-葡萄糖六号位氧原子上硫酸化, 那么你必须用6GA的残基, 而不是用0GA. 如果没有适合你用的残基的情况, 请看上面提到的注意事项.
- 如果想对酰胺基团上的N原子进行衍生化, 在乙酰基部分加上这些电荷. 相关的原子名称通常是这样的: C2N, O2N, CME, H1M, H2M, 和H3M. ich将会称为负值, 比如, 在GLYCAM的残基0YB上, 总和是0.106, 那么ich就是-0.106. 注意: 对于N-硫酸化, 对于每个连接变体, 电荷改变将是一样的, 比如, 0YB对6YB和WYB. 但是, beta-D-GlcpNAc相比alpha-D-GlcpNAc需要一种不同的改变, 等等.
- 找出dch和ich的差
- 比如, 当连接到氧原子上时, dch-ich=0.031. 所以, 你将需要将0.031加到糖环上每个与硫酸根相连的氧原子上.
- 对于使用beta-D-GlcpNAc, dch-ich=-0.057, 所以你需要把它加到连接的氮原子上.
制作拓扑前的准备:
由于AmberTools17中将GLYCAM 06h的相关参数移到了oldff文件夹下, 如果直接source或者load的话, 会出现不存在的状况, 所以:
mv $AMBERHOME/dat/leap/cmd/oldff/leaprc.GLYCAM_06h ..
mv $AMBERHOME/dat/leap/prep/oldff/GLYCAM_06h.prep ..
下面, 分三个教程详细叙述
创建Cell-O-的二糖拓扑文件
纤维素用碱处理之后, 在水溶液中, 羟基会变成氧负离子, 这种结构比较常用
新建一个cello.in的文件, 文件内容如下:
| cello.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 | # 加载参数
source leaprc.GLYCAM_06h
loadAmberPrep GLYCAM_06h.prep
# 定义多糖序列, 其中UGB的意思是在4号、6号位氧原子上连接有其它残基
cello = sequence{ ROH UGB }
# 设置将另一个葡萄糖残基连接在第一个葡萄糖环的4号位, 由于另一个我们也要在6号位碱化, 故设置为连接6GB
set cello tail cello.2.O4
cello = sequence{ cello 6GB }
# 根据教程中提到的, 需要改变碳原子的电荷
set cello.2.C6 charge -0.524
set cello.3.C6 charge -0.524
# 检查分子电荷是否为-2
charge cello
# 保存 top, crd, pdb文件
saveamberparm cello cello.top cello.crd
savepdb cello cello.pdb
# 退出
quit
|
创建甲基纤维素的拓扑和结构文件
残基-甲基的参数已经存在于GLYCAM_06h.prep中, 库中的名称为MEX, 使用时, 直接调用即可
新建一个mc.in的文件, 文件内容如下:
| mc.in | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 | # 加载参数
source leaprc.GLYCAM_06h
loadAmberPrep GLYCAM_06h.prep
# 定义多糖序列, 其中UGB的意思是在4号、6号位氧原子上连接有其它残基
mc = sequence{ ROH UGB }
# 设置将另一个葡萄糖残基连接在第一个葡萄糖环的4号位, 由于另一个我们也要在6号位甲基化, 故设置为连接6GB
set mc tail mc.2.O4
mc = sequence{ mc 6GB }
# 改变第一个糖环六号位上碳原子电荷, 将甲基连接到第一个糖环六号位氧原子上
set mc.2.C6 charge 0.243
set mc tail mc.2.O6
mc = sequence{ mc MEX }
# 改变第二个糖环六号位上碳原子电荷, 将甲基连接到第二个糖环六号位氧原子上
set mc.3.C6 charge 0.243
set mc tail mc.3.O6
mc = sequence{ mc MEX }
# 检查分子电荷是否为0
charge mc
# 保存保存 top, crd, pdb文件
saveamberparm mc mc.top mc.crd
savepdb mc mc.pdb
# 退出
quit
|
创建乙酰乙酸纤维素(CAA)的拓扑和结构文件
GLYCAM_06h.prep中, 只有甲基化和硫酸化的残基文件, 所以当需要其它衍生物时, 必须自己创建参数文件, 本教程在gaff力场下创建非标准残基, 后再连接该非标准残基
鉴于CAA的结构, 我们使用甲氧基作为封端基团, 创建一个小分子来计算电荷和制作非标准残基
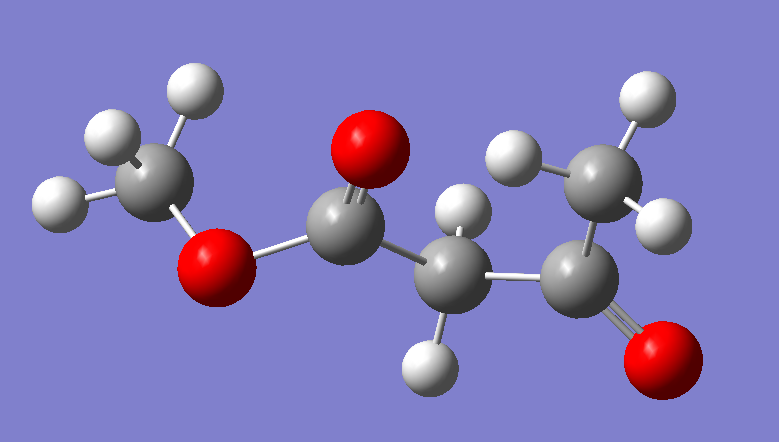
在GaussView中画好之后, 保存为高斯输入文件, 修改设定后, 文件内容如下所示:
| bash | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 | %mem=7GB
%nproc=4
%chk=CAA.chk
# hf/6-31g* opt freq pop=mk iop(6/33=2,6/42=6,6/50=1)
CAA
0 1
C -4.94088677 -3.40263880 0.11800459
C -5.35448344 -5.53099821 0.98523622
C -4.53967082 -5.86427353 2.24880574
C -5.05553271 -7.18031004 2.85998036
C -6.53361798 -7.57926158 2.69354239
O -4.74700696 -4.80462262 -0.08632844
O -6.55488107 -5.89787518 0.89569630
O -4.26925875 -7.92969947 3.49540093
H -5.66343006 -3.05738220 0.82767288
H -4.46510817 -2.70298085 -0.53698661
H -5.75663180 -3.46323083 -0.57176976
H -4.64754483 -5.07359443 2.96160990
H -3.50723724 -5.97212505 1.98928896
H -7.10329361 -7.18577669 3.50934832
H -6.90846953 -7.18317693 1.77294299
H -6.61607213 -8.64602474 2.68269390
CAA_ini.gesp
CAA.gesp
|
注: CAA.gesp后至少有两个空行, 否则高斯会报错, 加freq做频率分析, 看有没有虚频, 有的话说明优化不彻底, 需要修改参数, 例如opt=tight等, 由于原子数很少, 加上用的HF方法, 计算很快, 我们需要的是CAA.gesp文件, 它包含了静电势信息, 用于拟合resp电荷
拟合计算resp电荷, 并赋予原子在gaff力场中对应的原子类型参数
antechamber -i CAA.gesp -fi gesp -o CAA.ac -fo ac -pf y -c resp -at gaff
在生成的文件中, 你可以看到CAA.ac文件, 它有点像PDB文件, 但是包含了成键列表, 原子电荷和原子类型参数.
提醒: 你通常会使用antechamber来创建一个可以被LEaP读入的mol2文件. 由于随后我们需要使用prepgen处理生成的文件, 所以我们需要使用prepgen支持的ac格式的文件, 因为这是prepgen支持的唯一格式.
准备LEaP使用的残基库文件和力场参数
我们需要去除其头部的几个原子, 从而产生一个”氨基酸”式的残基, 该残基可以直接通过N-端和C-端与其他氨基酸连接.
(注: 是去除头部还是尾部的原子, 看你所画的分子中原子的顺序, 如有不明白的, 请参考Amber基础教程B5:模拟绿色荧光蛋白及构建修饰的氨基酸残基)
这部分可以通过使用prepgen程序配合mc(主链)文件完成, 该文件定义哪些原子需要被移除, 哪些原子是主链的一部分(例如骨架原子). 通常需要自己创建该文件. 我们创建一个CAA.mc的文件, 文件具体内容如下:
HEAD_NAME C2
MAIN_CHAIN C3
MAIN_CHAIN C4
MAIN_CHAIN C5
OMIT_NAME C1
OMIT_NAME H1
OMIT_NAME H2
OMIT_NAME H3
OMIT_NAME O1
PRE_HEAD_TYPE O
CHARGE 0.193333
HEAD_NAME和TAIL_NAME行的原子将会连接前后氨基酸残基. MAIN_CHAIN行列出连接头和尾原子之间的主链的原子, OMIT_NAME行列出了应该从CRO残基的最终结构中去除的原子, 因为它们不存在于完整的蛋白质(纤维素等多糖)中. PRE_HEAD_TYPE和POST_TAIL_TYPE行让prepgen知道周围蛋白质中的哪些原子类型将用于形成共价连接. (需要记住的是prepgen除了用于蛋白和多肽以外还可以用于其他的聚合物) CHARGE行指定了残基的总电荷, prepgen将确保”删除”原子的电荷在剩余的原子之间重新分配, 以便总电荷是正确的. (在这个例子中为0.193333, 得到这个数值的方法是在CAA.ac中把去除原子后剩余原子的电荷加起来)
prepgen -i CAA.ac -o CAA.prepin -m CAA.mc -rn CAA
运行该命令后你将会得到一个定义了CAA残基的CAA.prepin文件
现在, 我们有了自定义的的残基库文件, 它包含了我们需要的残基中各原子的电荷. 接下来我们需要检查其共价参数(键, 角和二面角)是否齐全. parmchk2程序将会检查哪些参数是需要的, 并且会在标准参数文件中寻找. 如果没有找到, 它将会尝试进行经验猜测, 并且将这些新的参数输出到一个我们命名为CAA.cro的文件中.
使用如下命令运行parmchk2(同样的可以使用-help选项查看所有可用的设置):
parmchk2 -i CAA.prepin -f prepi -o CAA.cro -a Y
完成这一步之后, 查看CAA.cro. 你或许会看到标记为ATTN, need revision, 参数都为0的行存在问题(本教程没有). 这表示parmchk2在gaff数据库中找不到适当的相似参数. 另外还有其他许多参数的选用是有高风险性的(这表明parmchk2预测的这些参数不太适合). 我们创建的CAA.cro文件如下:
| CAA.cro | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 | Remark line goes here
MASS
c 12.010 0.616
o 16.000 0.434
c3 12.010 0.878
hc 1.008 0.135
BOND
c -o 637.70 1.218
c -c3 313.00 1.524
c3-hc 330.60 1.097
ANGLE
c -c3-hc 46.930 108.770
c -c3-c 63.380 111.630
c3-c -o 67.400 123.200
c3-c -c3 62.040 116.500
hc-c3-hc 39.400 107.580
DIHE
o -c -c3-c 6 0.000 180.000 2.000
c3-c -c3-c 6 0.000 180.000 2.000
o -c -c3-hc 1 0.800 0.000 -1.000
o -c -c3-hc 1 0.000 0.000 -2.000
o -c -c3-hc 1 0.080 180.000 3.000
c3-c -c3-hc 6 0.000 180.000 2.000
IMPROPER
c3-c3-c -o 1.1 180.0 2.0 Using the default value
NONBON
c 1.9080 0.0860
o 1.6612 0.2100
c3 1.9080 0.1094
hc 1.4870 0.0157
|
创建多糖残基和衍生物残基连接处的一些键长、键角、二面角信息参数文件
由于我们使用的是GLYCAM和GAFF力场混用构建一个分子的参数, 而两个力场对同一个键定义的原子类型不同, 所以在两个残基连接处的一些参数需要自己去定义, 不然会出现找不到参数的错误(当然, 需要哪些参数, 得先进行下一步, 只不过loadAmberParams CAA_incomplete.frcmod就没有必要写了, 出现错误后就会知道需要哪些参数)
我们先给出这个文件, 后面再说明
| CAA_incomplete.frcmod | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | Remark line goes here
MASS
BOND
Os-c 450.00 1.323
ANGLE
Cg-Os-c 60.000 117.000
Os-c -o 80.000 125.000
Os-c -c3 95.000 110.800
DIHE
Cg-Cg-Os-c 1 -0.010 0.000 -3.000
Cg-Cg-Os-c 1 0.040 0.000 -2.000
Cg-Cg-Os-c 1 0.120 0.000 1.000
Cg-Os-c -o 1 0.000 0.000 -3.000
Cg-Os-c -o 1 -2.900 0.000 -2.000
Cg-Os-c -o 1 0.600 0.000 1.000
Cg-Os-c -c3 1 0.000 0.000 -3.000
Cg-Os-c -c3 1 -2.900 0.000 -2.000
Cg-Os-c -c3 1 -0.600 0.000 1.000
H1-Cg-Os-c 1 0.000 0.000 3.000
IMPROPER
NONBON
|
我们在$AMBERHOME/dat/leap/parm/GLYCAM_06h.dat中找提示中所见到的缺失的成键信息, 并将GAFF力场中的原子类型和GLYCAM力场中的原子类型比对, eg.GAFF力场里面的c对应着GLYCAM中的C, 所以, Os-c就是Os-C, 只需要在GLYCAM_06h.dat中找Os-C的参数写到CAA_incomplete.frcmod中就行了, 同理c3对应着Cg, 找到相应参数, 写入文件中即可, 特别注意的是, 二面角参数有个有两到三行参数, 则只需如上面看到的那样, 都写进去即可, 合理的解释可以看以下博文Amber力场二面角参数的解释.
创建用于模拟的拓扑和坐标文件
新建一个CAA.in的文件, 文件内容如下:
source leaprc.GLYCAM_06h
loadAmberPrep GLYCAM_06h.prep
source leaprc.gaff
loadAmberPrep CAA.prepin
loadAmberParams CAA.cro
loadAmberParams CAA_incomplete.frcmod
caa = sequence{ ROH UGB }
set caa tail caa.2.O4
caa = sequence{ caa 6GB }
set caa.2.C6 charge 0.282667
set caa tail caa.2.O6
caa = sequence{ caa CAA }
set caa.3.C6 charge 0.282667
set caa tail caa.3.O6
caa = sequence{ caa CAA }
charge caa
saveamberparm caa caa. top caa. crd
savepdb caa caa.pdb
quit
特别注明:
电荷的修改是至关重要的, 关系到生成的分子的总电荷是否符合实际, 本教程中0.282667的得出过程如上述所提到的那样, 衍生物残基的总电荷rch=0.193333, 衍生物期望的正式电荷为fch=0, dch=fch-rch=-0.193333, 而ich=-0.194, 那么碳原子需要的电荷改变量为: dch-ich=0.000667, 而碳原子原本的电荷为0.282, 所以需要修改碳原子电荷为: 0.282667.
