- 2019-07-14 16:11:39 许楠
AMBER 系的 GAFF 力场参数化有机小分子很有优势, 但是处理流程稍显复杂, 如图1.
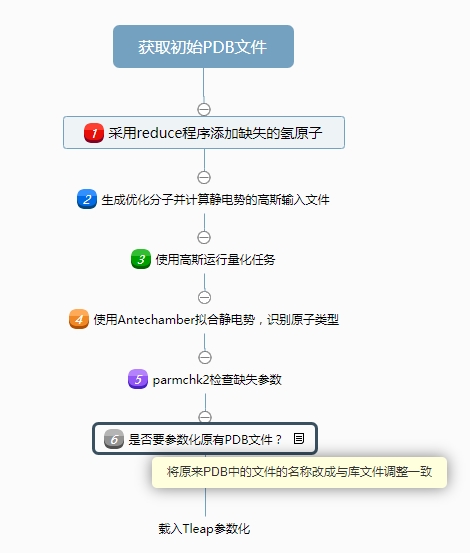
笔者开发了用于自动化处理小分子残基的前处理与后处理脚本, 可以方便地进行小分子参数化.
用户需要安装 Ambertools 套件和 gaussian 软件.
下面将以两个例子简要介绍脚本的使用.
示例1: 单分子, 使用GAFF力场参数化乙酸乙酯
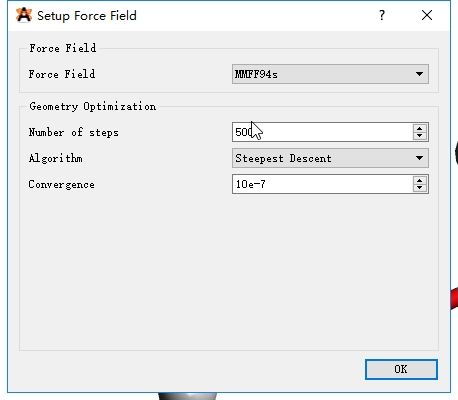
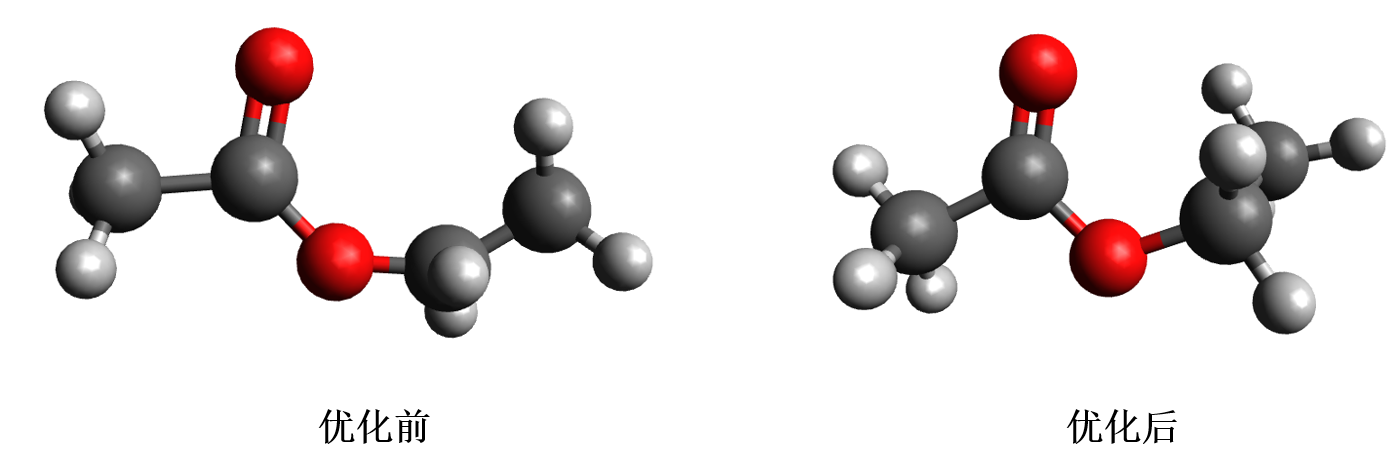
我们需要从头创建乙酸乙酯的坐标文件, 可以使用GaussView(收费)或者Avogadro(免费). 这里我们用免费的Avogadro. 首先选择 Draw Tools 模式(默认, F8 快捷键切换), 在屏幕上点击就会出现一个甲烷分子, 再从碳原子的位置点击并向旁边拖动光标就会自动出现乙烷分子, 坐标面板把元素调整为 O, 成键方式选择双键, 做类似的拖动操作就会出现一个乙醛分子. 再切换元素和成键方式绘出乙酸乙酯分子. 此时的结构显然不太合理, 我们可以使用Avogadro中的分子力场优化功能预优化分子结构. 依次点击菜单栏 Extensions-Molecular Mechanics-Setup Force Field, 选择 MMFF94s 力场, 然后点击 Extensions-Optimize Geometry 自动优化分子结构. 将优化后的结构导出为PDB文件 ea.pdb.
在Linux服务器上运行以下命令(需安装 Ambertools 套件):
python pre.py ea.pdb 0
Please specify a residue name for the molecule,3 capital letter-->ETA其中 ea.pdb 是PDB的名字, 0是分子的电荷. 程序会自动进行加氢操作, 本例中无任何氢原子丢失, 所以并未加任何氢原子. 接着程序会生程用于优化结构和静电势拟合的高斯输入 gjf 文件. 程序会提示给这个残基其一个三个大写字母的残基名, 这里叫它 ETA. gjf 文件生成后, 电荷, 残基名信息会保存在一个叫 RESNAME.txt 的文件夹内, (处理不同残基时不能混用!)
gjf 文件内容如下, nproc 是核心数, mem 是申请的内存数, 可以根据自己服务器的信息做适当修改, 但是内存不能超过服务器的物理内存. 计算静电势这里用的是比 HF/6-31G* gas MK 更好的 B3LYP/6-311G(d,p) D3BJ 隐式溶剂模拟 CHELPG 方法(参考 http://sobereva.com/441). 如果计算力不够, 可以考虑换成博文中的方法, 换成 def2SVP 优化再用 def2TZVP 基组计算静电势.
| ea.gjf | |
|---|---|
1 2 3 4 5 6 7 8 9 10 | %nproc=2
%chk=molecule
%mem=1GB
#B3LYP/6-311G** em=GD3BJ opt scrf=solvent=water iop(6/33=2) pop=CHELPG
remark line goes here
0 1
C -3.6000000000 2.1470000000 -0.0890000000
.... |
使用Gaussian计算完成后, 可以执行以下命令:
python post.py程序会自动调用Antechamber识别原子类型并拟合 RESP 电荷, 并输出在 LEaP 中载入该残基的语句. 最后程序还会调用 parmchk2 自动检查参数, 生成 ETA.frcmod 文件, 里面的力场参数是 parmchk2 根据半经验估算出来的, 需要我们判断是否合理.
Fintting RESP charge...
Checking parameters...
Tleap sentences for your reference.
source leaprc.gaff
set default PBRadii mbondi3
ETA= loadmol2 ETA_rename.mol2
loadAmberParams ETA.frcmod
No parameters are missing, but should be checked by yourself!我们在 Tleap 中加载这个残基就行了:
> source leaprc.gaff
> set default PBRadii mbondi3
> ETA= loadmol2 ETA_rename.mol2
Loading Mol2 file: ./ETA_rename.mol2
Reading MOLECULE named ETA
> loadAmberParams ETA.frcmod
Loading parameters: ./ETA.frcmod
Reading force field modification type file (frcmod)
Reading title:
Remark line goes here
> check ETA
Checking 'ETA'....
Checking parameters for unit 'ETA'.
Checking for bond parameters.
Checking for angle parameters.
Unit is OK.
> save ETA ETA.pdb至此乙酸乙酯残基已经参数化完成. 怎么生成系统的拓扑和坐标文件, 可以参考此博客内的其他教程http://jerkwin.github.io/.
示例2: 配体, 使用GAFF力场参数化HIV逆转录酶(HIV-RT)的抑制剂Sustiva
教程来源于 Amber基础教程B4-使用antechamber和GAFF模拟药物分子. Sustiva是小分子, 也是非标准残基, 没有与蛋白质有连接作用.
因为Sustiva是配体, 可以直接从RT-sustiva复合物的PDB文件(PDB编号: IFKO)中抽取出来的. 把 1fko.pdb 文件最后EFZ(Efavirenz 依法韦仑 )残基的原子提取出来(HETATM开头的行), 另存为EFZ.pdb. 接着执行与示例1一样的前处理, 后处理操作. 提示我们输入残基名时一定要与原来PDB中的一致, 也就是EFZ. 与示例1不同的是, 我们需要用加载好的EFZ残基参数化 1fko.pdb(因为其中还有尚未处理的非标准残基, 因此只加载含有sustiva分子的PDB, EFZ.pdb), 也就是最终sustiva分子的坐标是其与蛋白复合时的结构, 而不是高斯优化的结构.
参数化 EFZ.pdb 使用以下命令:
> source leaprc.gaff
> set default PBRadii mbondi3
> EFZ= loadmol2 EFZ_rename.mol2
> loadAmberParams EFZ.frcmod
> SUS=loadpdb EFZ.pdb
Loading PDB file: ./EFZ.pdb
total atoms in file: 30
> check SUS
Checking 'SUS'....
Checking parameters for unit 'SUS'.
Checking for bond parameters.
Checking for angle parameters.
Unit is OK.值得注意的是, 原来PDB中的分子加载加来时叫SUS, 这个不是残基名, 是这个单元的名字.
脚本和测试例子均可在我的Github上下载.
