- 2020-12-15 10:40:23
按: 本文主要汇编自网上的两篇文章:
- 王浩博: AlphaFold2
- Carlos Outeiral Rubiera: CASP14: what Google DeepMind’s AlphaFold 2 really achieved, and what it means for protein folding, biology and bioinformatics; 翻译: DeepL; 校对: 郝寅静, 惠成功; 统稿: 李继存
前言
1969年Cyrus Levinthal(塞莱斯·力文所)写到, 对一个蛋白质来说, 其构象空间可以高达$10^{300}$, 然而自然界中的蛋白质可以在微秒级别折叠, 这是一个悖论.
蛋白质结构预测是结构生物学一个里程碑式的问题, 每两年, 人类会组织一场蛋白质结构预测大赛, 即结构预测关键评估竞赛(CASP), 它堪称该领域的奥林匹克赛. CASP14的会议安排在2020年12月1号美东时间10点, 也就是北京时间, 周二的晚上11点, 世界上成绩最好的三支队伍将要给我们讲解他们今年在CASP14上创造了什么样的成绩?!

如果说两年前AlphaFold还是以A7D出道, 传统的课题组没有对他进行一点防备, 那么今年就完全不同了, 比赛前大家似乎卯足了劲, 工业界的队伍也多了不少, 从CASP14摘要上来看, 目前的工业界队伍(搜索.com粗略估计), 除了DeepMind的AlphaFold 2, 还有一些工业巨头, 比如微软的BrainFold, 微软亚洲研究院的两款算法, TOWER and FoldX 和 NOVA, 腾讯的数款算法tFold.
不过最值得一提的是一名上届程序未写完, 利用业余时间在Rust上实现PBuild算法的日本小哥, T. Oda. 遗憾的是这名小哥在摘要中提到了结构好像没有折叠出有意义的. 不过这种自由探索的精神可嘉, 这是真心爱科学, 希望他在未来继续努力!

(题外话, 他还让我想到了默默无闻在一家小公司开发质谱, 最后得了诺贝尔奖的田中耕一. 这些人的出现就是科研的土壤培养出来的. 如果我们各种普通人都有极大兴趣能够参与科学, 说明我们的土壤也肥沃起来了.)
让我们先来回顾一下, 之前的CASP都发生了什么?
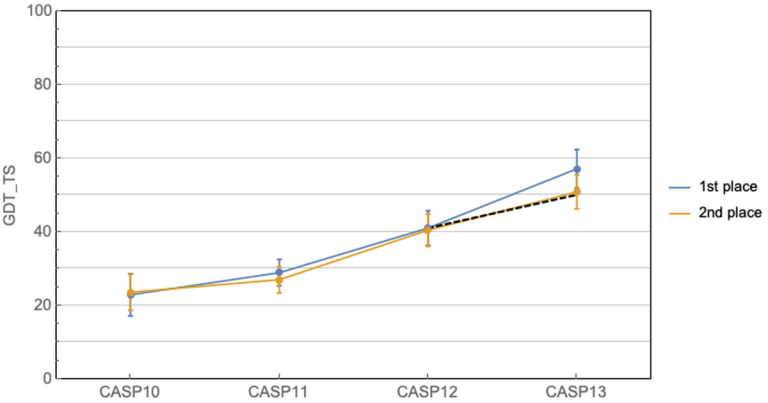
这几十年科学家们一直在缓慢而且努力的前进着. CASP11中协同进化方法开始崭露头角, 有了一个小的跳跃, 然后CASP12大家就齐头赶上了. 到CASP13的时候, AlphaFold来了, 利用深度学习结合协同进化的算法, 力压群雄, 碾压各种人类传统模型, 把第一名和第二名的差距直接拉大了!
我们来看一下今年CASP14的结果! Alphafold 2直接到了血虐其他算法的地步. 其实学术界的BAKER今年的进步也非常明显, 只不过在AlphaFold面前不值得一提了.
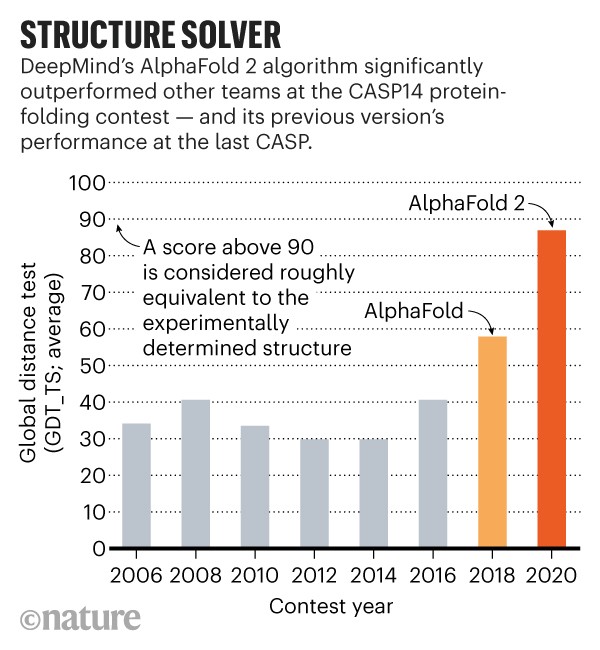
CASP用来衡量预测准确性的主要指标是GDT, 范围为0-100. 简单来说, GDT可以近似地认为是和实验结构相比, 成功预测在正确位置上的比例. 70分就是达到了同源建模的精度, 根据Moult教授的说法, 非正式地说, 大约90 分可以和实验结果相竞争!
这次DeepMind直接把总分干到了92.4, 和实验的误差在1.6埃, 即使是在最难的没有同源模板的蛋白质上面, 这个分数也达到了了恐怖的87.0.
来自牛津蛋白质信息学小组的报告
免责声明: 这篇博客是作者作为一名研究蛋白质建模的博士生基于参加CASP14会议过程传递的经历和观点. 提供的引用内容是从我的会议笔记中摘录的, 尽管我希望尽可能准确地记录这些内容, 仍无法保证它们是与会者所述的逐字转录. 牛津蛋白质信息学小组和我都不对这篇文章的内容承担任何责任.
你可能从科学或常规媒体, 甚至可能是DeepMind自己的博客上听到过, 谷歌的AlphaFold 2无可争议地在第14届结构预测关键评估竞赛CASP14夺冠——该竞赛是为计算生物学家预测几种蛋白质结构的两年一次的盲测; 这些蛋白质的结构已经通过实验确定——但尚未公开发布. AlphaFold 2的预测结果准确得令人难以置信, 以至于很多人已经欢呼这个代码就是长期存在的蛋白质结构预测问题的解决办法.
蛋白质结构是生物化学的核心, 对医学和技术有重大影响. 建立蛋白质结构是基于结构的药物设计的瓶颈, 准确的结构预测有望提高药物研究流程的生产力(尽管这只是一个因素, 在真正的革命性变化发生之前, 我们还需要把其他事情做好——请在这里和这里查看Derek Lowe的文章). 蛋白质的结构信息在生物学中也是至关重要的, 它有助于阐明功能——生物化学中的许多关键论文都从结构测定的实验进展中获得洞见.
鉴于这个问题是如此地重要, 以及几十年来网络资源的缓慢发展, 我想没有人想到解决方案会很快出现. 我自己也决定将博士研究的重点放在结构预测领域, 我和很多人一样认为, 在我们能够取得接近解决方案的成果之前, 需要进行几年工作, 跨越很多研究线. 现在我可能需要换个课题了.
新闻稿中有多少是真实的, 实际发生了什么, 有多大意义? 关于这个话题, 在多个论坛上已经出现了无尽的讨论. 坦率地说, 在过去的72小时里, 我一直无法思考其他事情. 为了理清自己的思路, 我决定写下这篇博文, 详细介绍自格林尼治标准时间周一下午3点左右我的科学世界被颠覆后所了解到的一切. 我希望这对那些不能参加CASP14的蛋白质生物信息学家朋友们有用, 同时也对任何想多听一点这个话题的人有用.
请记住, 我对CASP14评估和会议的报告必然会夹杂着猜测. AlphaFold 2的工作细节仍然未知, 在他们的论文通过同行评审之前(根据他们的CASP13论文, 这可能需要一年以上的时间), 我们可能无法完整了解这些细节. 突破程度是不可否认的——但我们需要更多的细节来衡量其潜在的影响.
这将是一篇很长的文章. 不要说我没有警告过你.
AlphaFold 2到底有多好?
惊人得好.
CASP14组织者, John Moult , 一位近70岁, 伦敦口音的老绅士在这届会议上娓娓道来, I wasn’t sure that I would live long enough to see this.
结构生物学家Osnat Herzberg: 预测的结果好像和我做的结构不大一样, 咦?我怎么解错了.
结构生物学家Petr Leiman: 我用着价值一千万美元的电镜, 还这么努力的解了好几年, 这就一下就给我算出来了??
让我来告诉你发生在上周一的事. 在CASP14会议开始前的几个小时——格林尼治标准时间中午左右——主办方公布了评估结果. 马上, 评论开始在Twitter上流传. 这是大家都在分享的图片:
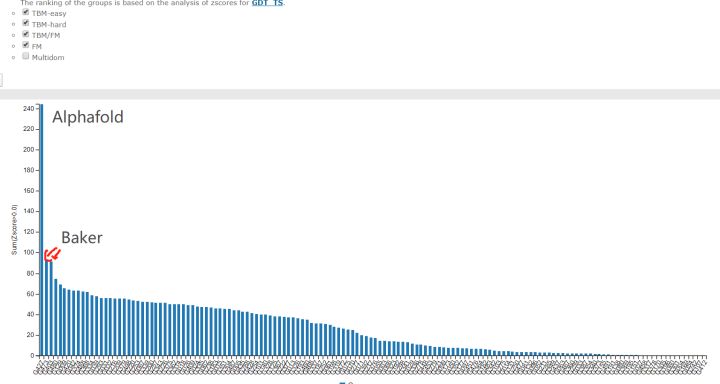
综合成绩
最难的free modelling部分
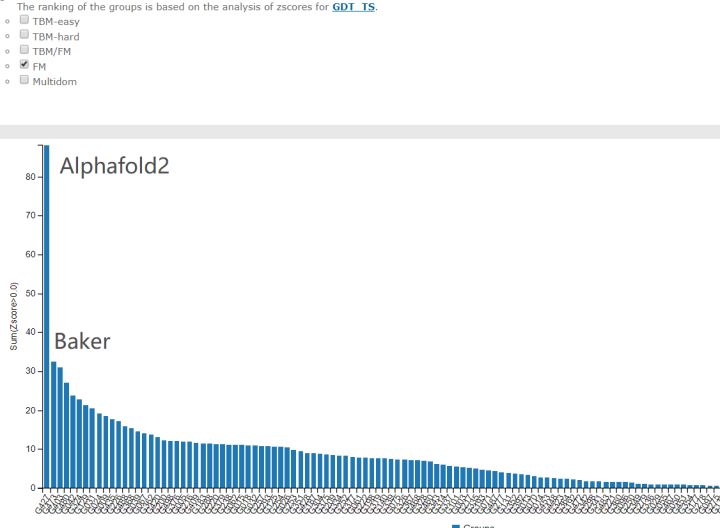
根据预测的Z-score总和(如果大于零), 对CASP14参与者进行排名. 第一名AlphaFold 2(427组)相对于第二名BAKER(473组)表现出了惊人的提高. 此图来自2020年12月1日(星期二)CASP14官方网页.
这个柱状图给出了不同组的预测Z-score之和. 请记住, Z-score只是样本值相对于群体平均值的差, 然后除以标准差所得的值; 数值越高, 代表与平均值的偏差越大, 这是一种常用的离群值检测方式. 换句话说, 明显优于平均水平的组, 其Z-score会更大. 在这张图中, 我们看到有一个组的表现远远好于其他组. 当考虑所有目标时, 427组的平均Z-score大约2.5, 而在最困难目标中, 其Z-score上升到3.8. 如果这是一个智力测试, AlphaFold 2的智商(IQ)会超过160分.
如果说相对比较令人震惊, 那么实际表现也同样令人印象深刻. 我将考虑结构生物学中的一个典型指标, 原子位置的均方根偏差(RMSD). 如果你不是很了解蛋白质折叠, 这些数字对你来说可能没太多意义. 别担心——在下一节中, 我将展示一些图形化的例子. 只要记住: (1) 较低的RMSD代表着更好的预测结构; (2) 大多数实验结构的分辨率在2.5 Å左右(12月8日更新: 尽管, 正如许多人在Twitter上指出的那样, 这种比较并不恰当). 考虑到这一点, 427组提交的目标中, 约有三分之一(36%)的预测均方根偏差(RMSD)低于2 Å, 86%低于5 Å, 总平均值为3.8 Å.
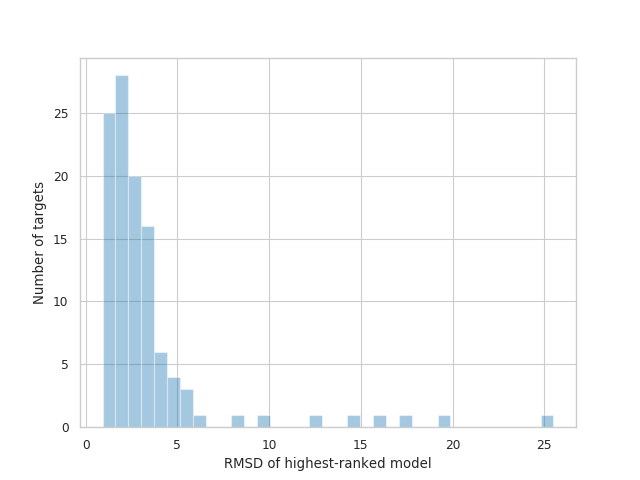
AlphaFold 2提交的排名最高的模型的RMSD分布. 数据来自2020年12月1日(星期二)CASP14官方网页.
当会议开始时, 我们仍然在消化这些信息, 哦, 天哪, 前半小时对我们简直是折磨. 先是声称今年的比赛”有点不寻常”, 随后提到一个特定的小组已经得到了令人印象深刻的结果. 最后, 自1994年以来一直担任每届CASP主席的John Moult, 熟练地对比赛的历史作了一番扣人心弦的阐述, 慢慢地给我们提供信息, 直到他最终展示了我们都期待的图表, 也就是下图:
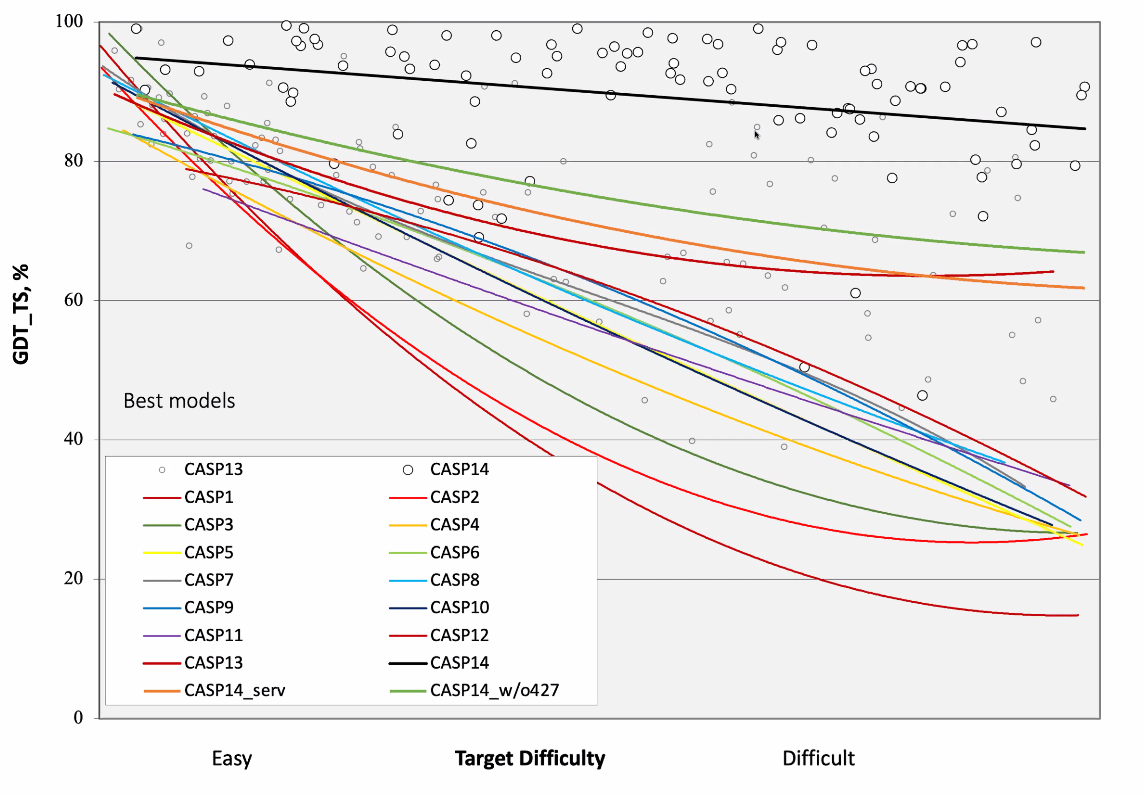
历届CASP竞赛的综合结果. 深橙色的线(CASP14_serv)对应的是全自动服务器给出的预测, 橄榄绿色的线(CASP14_w/o427)包括除表现最好的小组之外, 所有由人工辅助的预测, 黑色的线(CASP14)代表表现最好的小组, 427组, 即AlphaFold 2的预测. 该图使用GDT_TS评分, 其中100代表完美结果, 0表示无意义的预测.
根据经验判断, GDT_TS在60%左右代表”正确折叠”, 意味着我们对蛋白质如何整体折叠有点概念; 而超过80%则表示侧链与模型非常接近. 正如你所看到的, 除了一小部分任务外, AlphaFold 2在其他任务中都达到了这个目标.
然后, 经过三十年的竞争, 评估人员宣布AlphaFold 2成功地解决了一个公开了50年的挑战: 开发一种方法, 能够准确地, 普遍地, 有竞争力地从蛋白质序列(或者, 好吧, 我们将在后面看到是多序列比对)预测蛋白质结构. 同其它任何应用一样, 现在还存在着一些注意事项和边缘情况——但这一突破的规模及其潜在影响不可否认.
故事并没有到此结束. AlphaFold 2产生的模型非常好, 以致于在某些情况下与实验结果相悖. 我将根据会议中提到的例子举两个简单的例子. 第一个例子来自Osnat Herzberg组, 他们正在研究一种噬菌体尾丝蛋白. 在看到DeepMind预测的模型与他们的结构有很好的一致性后, 他们注意到他们对顺式脯氨酸有不同的指定. 在重新回顾了分析之后, 他们意识到自己在解读结构时犯了一个错误, 并进行了纠正.
第二个例子来自Henning Tidow组, 他正在研究一种整合膜蛋白——还原酶FoxB(看起来与革兰氏阴性菌的铁吸收有关). Tidow组对这个模型进行了大约两年的研究, 尝试了不同的方法来获得晶体结构, 包括实验性的相位方法. 当他们得到DeepMind预测的模型后, 他们在几个小时内就成功地通过分子替换解决了问题.
最后还有一点需要说明. 有人曾怀疑, 谷歌不可思议的成功是否与今年的这组目标蛋白更容易没有关系. 这个说法本身就很难成立(毕竟其他经验更丰富的小组不也会因此受益吗?), 但为了反驳这一点, 评估人员根据现有蛋白结构与目标的相似性, 得出了CASP14的任务目标是历届最难的:
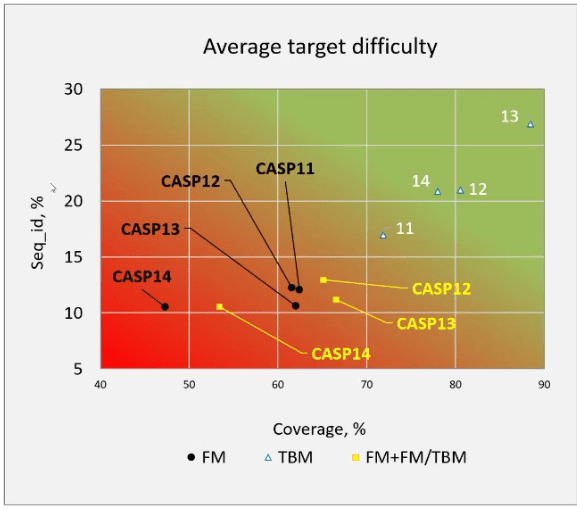
根据现有模板的覆盖范围和序列一致性对最后四届CASP目标的比较. 在这两方面, CASP14包括了迄今为止提供过的最难的自由建模(FM)目标. TBM代表基于模板的模型.
还有有许多有趣的讨论点. 许多人会认为, CASP14研究目标的集合并不能代表 所有 有趣的结构预测问题. 他们会是对的. 是的, 当然有一些问题, AlphaFold 2的表现并没有那么好. 在这篇博文快结束的时候, 我会讲讲自己对一些注意事项的看法. 但是, 现在我们要明确一点: AlphaFold 2是一个可以解决 相当多目标蛋白质 结构预测的工具.
与其他方法相比如何?
我可能已经说服了你, AlphaFold 2是一个巨大的突破. 现在是时候从评估者发现了什么这一坚实基础, 沿着越来越多的猜测向下探讨其他小组是如何做的, 做了什么, 然后是AlphaFold 2的方法, 然后去预测这对生物学, 特别是生物信息学可能意味着什么.
我将对竞赛中的两个目标进行仔细的, 尽管是简短的考察, 将AlphaFold 2与两个排名最好的小组, David Baker组和Yang Zhang组进行比较. 他们 (1) 在过去的CASP比赛中一直表现非常好, (2) 在本周二发表了精彩的演讲, 所以我对预测背后的情况有不错的了解.
我准备考察的第一个目标是ORF8蛋白, 这是一种参与SARS-CoV-2和免疫反应之间相互作用的病毒蛋白(PDB: 7JTL, bioRxiv上有预印本). 在CASP14中, 它被标记为T1064. 让我们来看看AlphaFold 2预测的结构(红色)与晶体结构(蓝色)的比较:
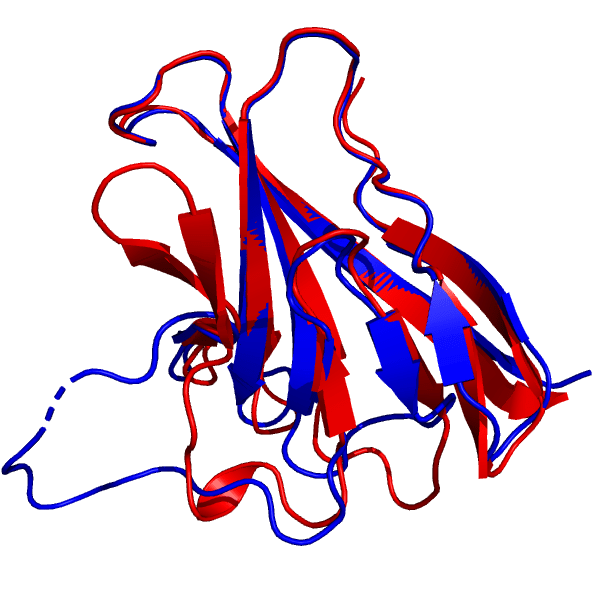
427组给出的T1064目标蛋白的最佳模型(红色), 叠加到7JTL_A结构上(蓝色). DeepMind的结构来自2020年12月1日(星期二)CASP14官方网页.
蛋白质核心的预测与实验非常一致, 完整再现了反平行β片层的结构, 更令人印象深刻的是连接它们的环形结构. 请记住, 环区的特点是缺乏二级结构, 意味着不像α-螺旋和β-片层那样, 有一个氢键的骨架将结构维持在一起. 由于这个原因, 一般认为环区很难预测, 与通常的方法相比, AlphaFold 2的表现相当惊人.
然而, 请注意, 在图像的左下角, 有一个大的环形区域, 与晶体结构有显著区别. 除了环形结构的整体形状, 氢键模式也明显错误, PyMOL将其中相当一部分识别为β片层. 虽然这个含30个残基的环状区域位置错误, 但模型确实与环形区域有一定的相似性, 其表现仍优于大多数常用方法. 更重要的是, 由于环状区域通常柔性很大, 程序的失败可能只是指出这个区域是非固定的(在距离预测模型中有时会发生). 此外, 即使结构出错的百分比有两位数, 整体RMSD也只超过1 Å一点点.
12月10日更新: 来自Institut de Biologie et Chimie des Protéines的Juliette Martin向我指出SARS-CoV2 ORF8蛋白的一个新结构, 其中被AlphaFold 2预测错误的环形结构实际上与预测结构非常相似! 即使我们认为它失败了, 它居然也能做对.
其他小组的表现如何? Baker组和Zhang组都使用了类似的流程, 其中融合了CASP13 AlphaFold的许多想法: 建立一个多序列比对, 可能会考虑宏基因组学序列; 使用深度学习预测一个潜在的结构, 并使用他们实验室的特有方法找到一个最佳结构(Baker组使用ROSETTA, Zhang组使用I-TASSER), 并进行一些精修, 可能也是使用深度学习. 我不会深入讨论细节——请在CASP14的特刊中寻找他们的论文——相反, 让我们看看他们的表现(绿色为Baker组, 黑色为Zhang组).
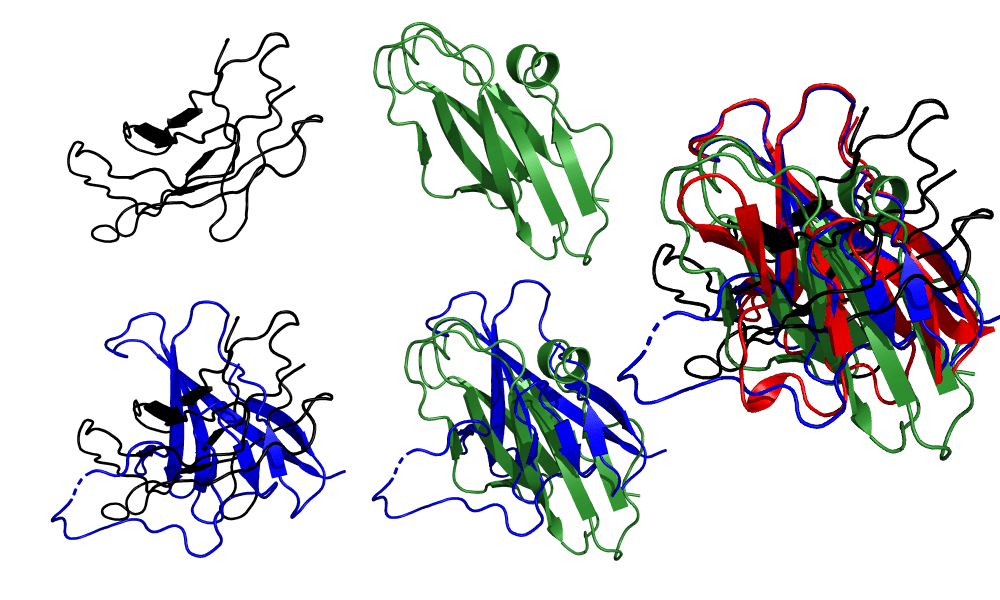
顶部: Zhang(黑色)和Baker(绿色)人工组提交的T1064目标蛋白的最高得分模型. 底部: 与晶体结构对齐后的模型. 右图: 所有三个模型(Zhang, Baker和AlphaFold 2)与晶体结构对齐. 图来自2020年12月1日(星期二)CASP14官方网页.
我们可以看到模型和晶体结构之间的明显差异. 两种模型都把核心的拓扑结构弄错了: Baker组的β片层比晶体结构多, 拓扑结构也是错误的, 把平行片层和反平行片层结合在一起了; Zhang组勉强抓住了核心的结构. 在这两种情况下, 连接β片层的环形区域到处都是, 对于AlphaFold 2没有正确建模的含30个残基的环形区域, 这两个组得到的结果更糟糕.
不要误会我的意思——这是一个困难的目标, Baker和Zhang的工作已经非常出色, 他们的预测在其他任何CASP中都会是最先进的. 这个目标的次佳的模型, 来自清华的Xianming Pan, 只是稍微好一点. 但有一点很清楚: 当与蛋白质结构预测界表现最好的小组进行比较时, AlphaFold 2的准确率简直是在一个完全不同的水平上.
虽然ORF8蛋白肯定不是他们模型中最差的, 但在DeepMind的演讲中, ORF8蛋白被强调为”他们做得不是那么好”的目标之一. 好吧, 让我们来看看他们实际上做得非常好的一些模型. 我们现在要看的是目标T1046s1(PDB: 6x6o, 链A).
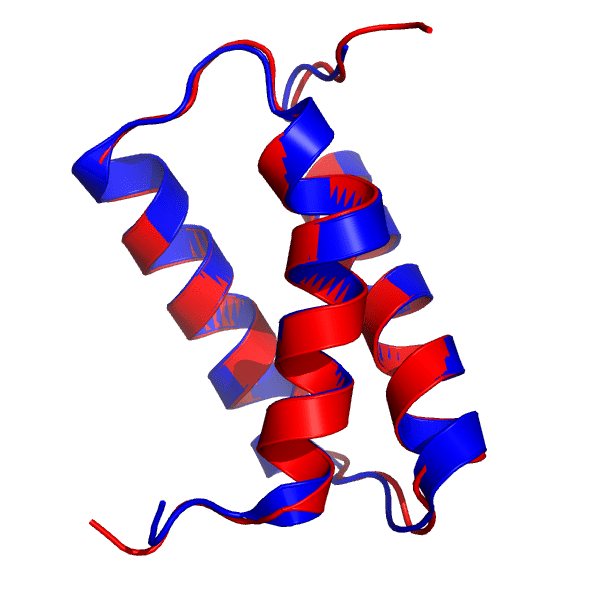
427组T1046s1目标蛋白的最佳模型(红色), 叠加在6X6O_A结构上(蓝色). DeepMind的结构来自2020年12月1日(星期二)CASP14官方网页.
在这里, AlphaFold模型与晶体结构几乎没有区别, 总的RMSD为0.48 Å. α-螺旋的精度简直完美, 特别是第一个α-螺旋(3D图像上最接近观察者的那个)的扭结以出色的精度再现. 与之前的目标一样, 连接主要二级结构部分的无规区域与晶体结构几乎无法区分. 唯一显示出明显差异的区域是N-和C-末端, 而且这些区域确实非常小.
由于这是一种相对简单的蛋白质(一种小型的全α螺旋蛋白), 所以并不奇怪, Baker和Zhang组建立的模型都能准确地重现折叠:
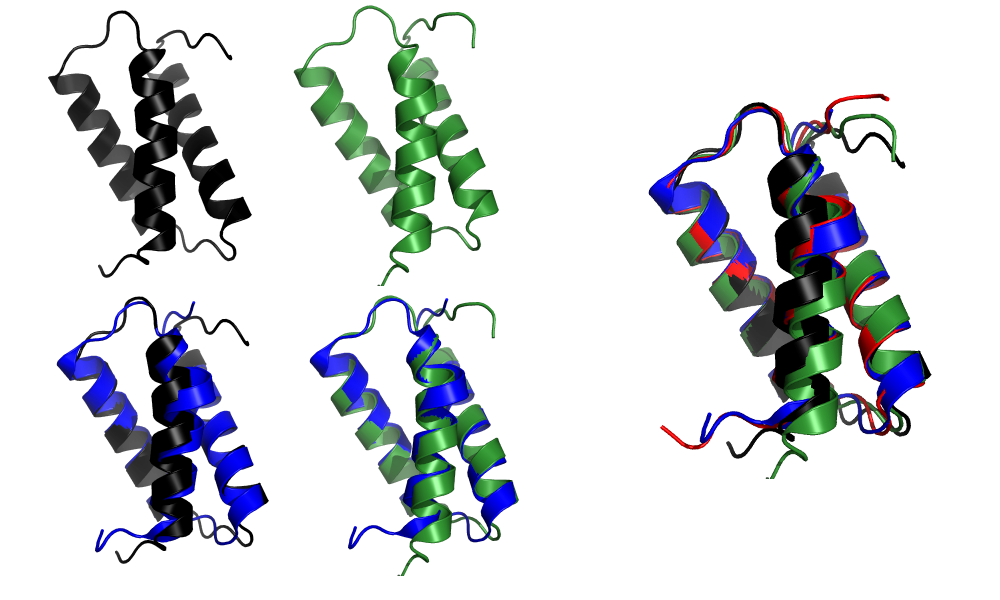
上图: Zhang(黑色)和Baker(绿色)人工组提交的T1046s1目标蛋白的最高得分模型. 底部: 与晶体结构对齐后的模型(蓝色). 右: 所有三个模型(Zhang, Baker和AlphaFold 2, 红色)与晶体结构(蓝色)的对齐. 参赛作品来自2020年12月1日(星期二)CASP14官方网页.
这些都是非常好的模型, 尤其是对连接α-螺旋的环形结构表现非常出色. 然而, 仔细检查后发现了一些差异. 第一个α螺旋的扭结没有准确地再现: Zhang组将其建模为基本上直的螺旋, 而Baker组则显示出较小的扭结; 相比之下, AlphaFold 2的扭结得非常准确. 而且, 这两个组模型偏差的幅度要比AlphaFold 2模型中大很多.
评测人员之一的Nick Grishin用一句话总结了这种不可思议的表现, 这句话大致是这样的: “AlphaFold 2做对了而其他模型没有做对的, 是什么? 细节”. 事实上, 模型符合得非常好, 甚至对侧链也是这样:
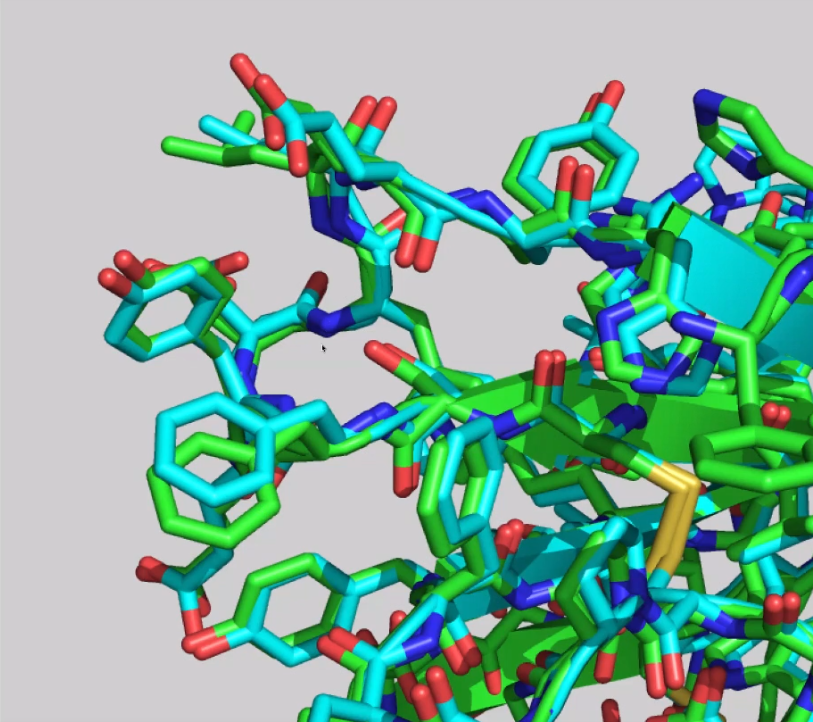
AlphaFold 2不仅能高度准确地预测蛋白质的全局结构, 还能对侧链结构做出令人难以置信的准确预测. 图片取自CASP14的幻灯片.
他们是怎么做到的? 第一部分: 技术细节
这将是一个困难的问题. DeepMind在CASP14摘要集中对他们所用流程的描述没有提供太多细节, 尽管他们的演讲确实提供了一些有趣的信息, 但很多东西还是未知的. 在他们发布相应的论文之前, 我们不会知道他们到底做了什么, 论文发表需要几个月甚至一年以上的时间. 不过, 我可以告诉你目前他们说了些什么, 我们可以试着猜测一下他们的底细.
首先看看DeepMind官网的说明:
A folded protein can be thought of as a “spatial graph”, where residues are the nodes and edges connect the residues in close proximity. This graph is important for understanding the physical interactions within proteins, as well as their evolutionary history. For the latest version of AlphaFold, used at CASP14, we created an attention-based neural network system, trained end-to-end, that attempts to interpret the structure of this graph, while reasoning over the implicit graph that it’s building. It uses evolutionarily related sequences, multiple sequence alignment (MSA), and a representation of amino acid residue pairs to refine this graph.
大致是说, 折叠的蛋白质可以视为”空间图”, 其中残基为结点, 边将残基紧密相连. 该图对于理解蛋白质内的物理相互作用及其进化历史非常重要. 对于在CASP14上使用的最新版本的AlphaFold, 我们创建了一个基于注意力的神经网络系统, 该系统经过端到端训练, 试图解释该图的结构, 同时对所构建的隐式图进行推理. 它使用进化相关序列, 多序列比对(MSA)和氨基酸残基对表示来完善此图.
此外, 里面没有提到深度增强学习, 倒是讲了目前学术界和工业界都非常关注的注意力模型. 同时, 用了128 TPUv3核心和几周的时间, 训练了PDB库中的约17万个蛋白. 这套模型需要几天的时间来预测一个高精度的蛋白质结构.
代表DeepMind参加会议的John Jumper报告时开篇讲了一下核心观点, 物理直觉融入到了网络结构中, 端对端直接生成结构取代了残基的距离矩阵, 从图的角度出发直接反映蛋白质的物理结构和几何.
John提到了数据库, 说是标准的数据库, 如序列库UniRef90, BFD, MGnigy Clusters, 结构库PDB, PDB70.
训练流程基本上和官网的图一致, 从序列出发得到MSA和template, 然后给了一个双线的transformer(似乎是在序列维度上和残基维度上分别做了softmax), 然后两者怎么交互的信息没怎么看明白, 不过看迭代边和序列的方式, 应该就是经典的GNN, 参考Deepmind的GNN, Transformer教程).
然后结合3D-equivariant transformer做端对端的训练, 训练完之后用amber优化一下. 参考Max Welling的3D Roto-Translation Equivariant transformer, 听说他们知道AlphaFold用了这个还挺吃惊. 听说这里有一个可微分的问题, 且需要neighborhood来帮助解决旋转平移不变性特别吃内存, 具体还不是很清楚.
这里没有预训练模型, 没有深度增强学习, 输入是MSA, 没有MRF/协同进化/precision matrix作为feature, 没有distance matrix作为最终的输出, 直接输出PDB, 整个框架都变了!
整个报告信息量不足, 这里给出的都是碎片化的相关信息. 至于AlphaFold的核心武器在哪里, 讨论下来, 大家把更多的目光放在了后面的实现端对端的3D-equivariant transformer上. 猜测端对端减少了embed协同进化信号带来的噪音, distance matrix只有主链信息, PDB结构可以提供额外的约束信息, 可以直接把模型质量反馈给前端的transformer.
这样看来, AlphaFold 2和大多数现代预测算法一样, 依赖多序列比对(MSA). 我们打算预测其结构的蛋白质的序列会在一个大型数据库中进行比对(通常是类似UniRef的数据库, 尽管在后来的几年里, 用源于宏基因组学的序列来丰富这些比对变得很常见). 其基本思想是, 如果两个氨基酸紧密接触, 其中一个氨基酸的突变将导致另一个氨基酸的突变, 这样才能保持结构.
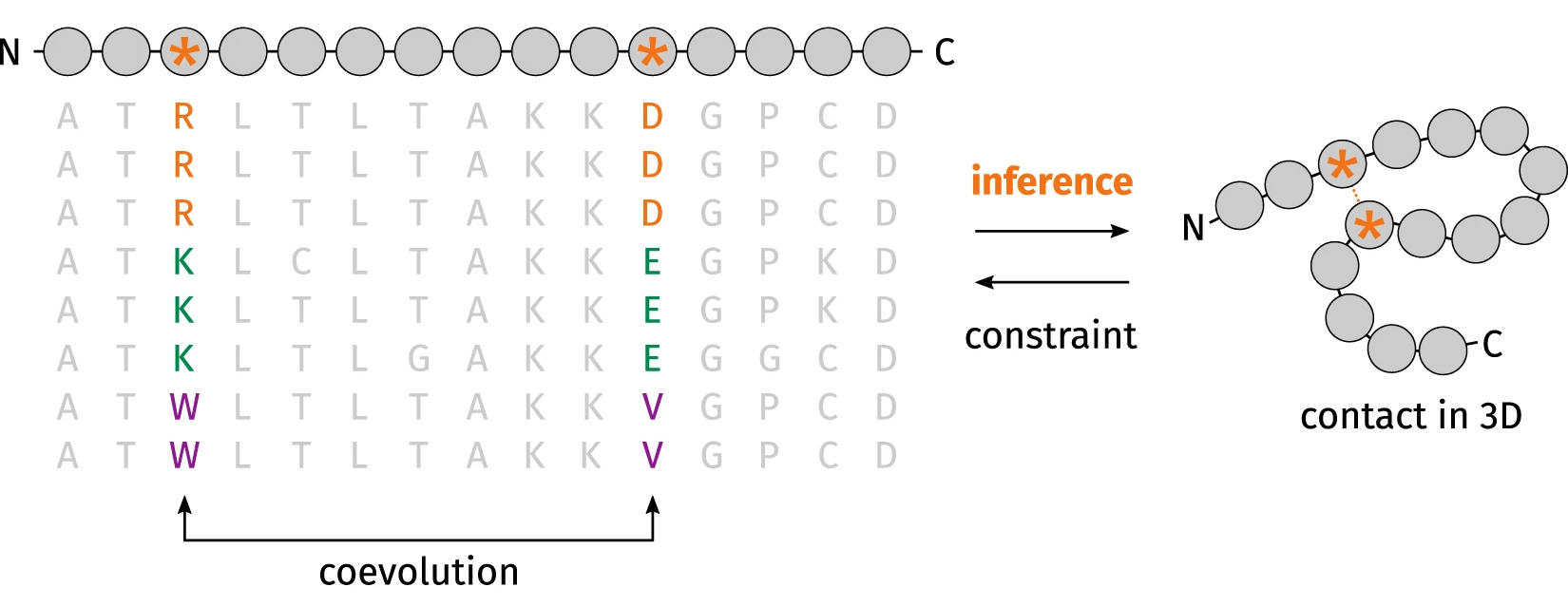
协同进化方法如何从多序列比对(MSA)中提取蛋白质结构信息的示意图. 图片修改自doi: 10.5281/zenodo.1405369.
考虑下面的例子. 假设我们有一个蛋白质, 其中一个带负电荷的氨基酸(如谷氨酸)靠近一个带正电荷的氨基酸(如赖氨酸), 尽管它们在氨基酸序列中都相距甚远. 这种库仑相互作用稳定了蛋白质的结构. 现在想象一下, 第一个氨基酸突变成一个带正电荷的氨基酸——为了保持这种接触, 第二个氨基酸将在进化压力下突变成一个带负电荷的氨基酸, 否则产生的蛋白质可能无法折叠. 当然, 真实的情况很少像这个例子一样清晰, 但你明白我的意思.
这个原理启发了非常多的算法来预测蛋白质的结构特性, 从接触到二级结构. AlphaFold自己在CASP13中的成功确实根据MSA(以及很多其他特征, 包括一些协同进化软件的输出)利用了深度学习来预测残基间距离. 然后, 这些预测将被转化为一个潜在的结构, 并进行能量最小化(使用一个简单的梯度下降算法, 如L-BFGS)以找到一个好的结构. 在CASP14中很多研究小组采用了这个想法, 包括非常优秀的一些研究小组.
但这次, DeepMind决定开发一个端到端的模型. 他们没有使用MSA来预测约束, 而是设计了一个深度学习架构, 将MSA作为输入(加上一些模板信息, 但这是另一个故事), 并在最后输出一个完整的结构. 他们的动机很简单. 鉴于PDB中可用的约17万个结构构成了一个小的数据集, 他们希望充分利用归纳偏向——在模型架构中引入约束, 确保信息被快速高效地同化.
为了理解DeepMind团队的目的, 让我们先考虑一下卷积神经网络的案例, 它是计算机视觉领域许多成功案例背后的深度学习架构. 许多人认为CNN的成功是由于它们限制信息流动的方式: 由于它们的设计, 一个像素所对应的信息与其邻居混合在一起, 这种局部性在各层中流动, 以分层的方式从不同区域提取信息 . 网络不需要使用大量的数据或训练时间来学习局部信息是重要的——相反, 由于架构所施加的约束, 这些信息会自然学习到.
他们尚不清楚他们如何使用归纳偏向. 我们知道, 输入信息被嵌入到一个嵌入空间中, 对于这个空间, 我们并没有太多的信息. 代表DeepMind参加会议的John Jumper解释说, 它”学习序列-残基边和残基-残基边”, 并提到该模型”采用了一个基于注意力的网络来识别哪些边是重要的”(相比之下, 原来的AlphaFold对所有距离的权重相等). 虽然我们对实际架构的信息不多, 但我们知道, 一个重要的部分是3D等价转变器(3D equivariant transformer)——这是一种新型的深度学习架构, 因其在GPT-3和BERT等著名模型中的作用而广为人知——它负责更新蛋白质骨架和构建侧链.
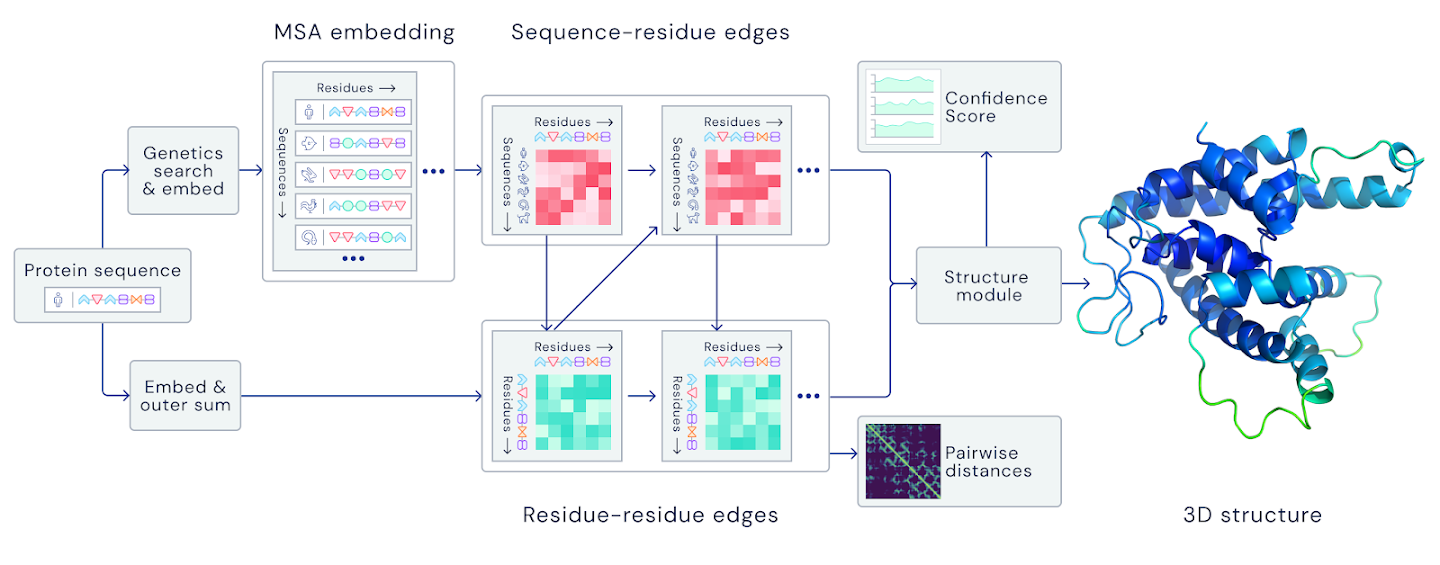
DeepMind的图(摘自他们的博客)提供了AlphaFold 2的架构概述, 但缺乏重现它所需的细节.
预测过程以迭代的方式进行, “在MSA和蛋白质的内部表示之间来回传递信息”. 我猜测这意味着从网络的正向传递中获得的信息会以某种方式反馈到输入特征中, 然后重新运行, 直到收敛——但这当然是一种猜测. 从大会上展示的图来看, 第一次的预测结果往往非常好(大约70-80 GDT_TS), 经过几次迭代之后, 就会收敛到我们在CASP14中看到的令人印象深刻的90+ GDT_TS的预测结果. 最终的结果并不能保证符合所有的立体化学约束, 所以使用Amber ff99SB力场和OpenMM通过坐标限制梯度下降来弛豫最终的结构.
当然, 没有足够的信息来尝试创建一个类似的模型. 我怀疑蛋白质信息学界的其他成员正在经历一场科学的悬念, 以比《赛博朋克2077》更高的热情等待DeepMind的论文. 同时, 我们也不清楚要朝哪个方向努力.
他们是怎么做到的? 第二部分: 不那么技术性的细节
当然, DeepMind团队的成功不仅与深度学习有关. 还有更多, 很多.
很多因素让人想起Mohammed AlQuraishi在上届CASP后的那个著名说辞——DeepMind组织了一个灵活的, 资金充裕的小组, 可以快速尝试很多想法, 交流信息的速度也比学术小组快得多, 他们每两年才交流一次. 我不想讨论这个问题, 因为我期待AlQuraishi在这次CASP之后会写一篇类似的文章(12月8日更新: Mo的文章). 相反, 我想讨论两个问题, 我认为这两个问题不仅对理解他们的成功很重要, 而且对考虑这种成功将如何影响学术计算研究也很重要: (1) DeepMind几乎无限的算力的影响; (2) 学术研究团体所产生和发表的大量结构和方法数据的影响.
我们先来谈谈计算资源. 当John Moult在介绍AlphaFold 2令人印象深刻的性能时, 第一批新闻稿也开始出现了, 有一个话题似乎占据了CASP14 Discord频道的主导地位: 有多少资源用于训练这个模型. DeepMind的博客文章指出, 他们的模型
使用了大约128个TPUv3核心(大致相当于约100-200个GPU), 运行了几周, 在当今机器学习中使用的大多数大型最先进模型的背景下, 这是一个相对适中的计算量.
AlphaFold: 解决生物学领域50年来的大挑战, 见DeepMind的博客.
张量处理单元(TPU)是谷歌开发的一种专有的专用集成电路(ASIC), 用于加速神经网络的训练. 与最初被设想为处理图形之后被转换用途的GPU不同, TPU从一开始就是为深度学习而设计的, 在DeepMind最近的大部分成功案例中都有它们的身影.
TPU和GPU之间并不存在明确的等价关系(就像GPU和CPU之间不存在一样), 因为性能取决于问题, 但正确使用时, 它们可以提供相当大的速度提升. 也许更重要的是, 一个8核TPU v3芯片拥有128GB的vRAM, 这对于一些内存成本较高的架构——比如注意力模型——是必要的. 仅供参考, 我所知道的具有最大内存的GPU是NVIDIA A100, 有40GB(虽然最近公布了这款GPU的80GB版本). 这是相当大的差距.
如果你觉得GPU很贵, 可以考虑一下, 按照谷歌云的定价页面, 租用128个TPUv2核心的年成本是50万美元. 使用云服务复制DeepMind的实验副本, 根据条件不同, 需要2.5万美元到20万美元不等——当然, 这还没有算上探索架构, 调试, 优化超参数或运行多个副本的计算工作量. 总的计算成本可能在几百万美元左右.
这一切都很好, 但是, 与其他参与者相比如何呢? 在一次问答中, Baker和Zhang小组表示, 他们大致用了4个GPU来训练自己的模型, 花了几个星期. 这意味着DeepMind团队的计算资源大概多了两个数量级. 当然, 像我们在前一段中估计的数字, 即使是资助最雄厚的计算研究小组也无法做到.
这种庞大的计算能力是DeepMind成功背后的唯一因素吗? 我不这么认为. 这个天才团队的工作展示了新颖的想法和创造性的问题解决方式, 而这些差异不能仅仅归因于处理器的强悍. 当然, 也不能忽视. 庞大的计算能力不仅意味着他们可以处理更大的模型——他们还可以实现比任何学术小组更高的吞吐量. Baker小组需要一个月的时间在他们的4个Titan GPU中测试的东西, 对于DeepMind来说可能只需要几个小时, 这使得快速原型设计和测试想法成为可能. 当然, 像最终形成AlphaFold 2的架构这样的想法, 在没有合适的硬件的情况下根本不会考虑.
展望未来, 人们不禁要问, 这种资源的不平衡将如何影响学术计算研究. 模型变得更大, 更复杂是一个明显的趋势, 而这一趋势的发生速度远远超过了硬件价格的下降速度. 除非我们能想出一种方法来改善对快速增加的计算资源的需求, 而价格又能承受, 否则我们可能最终会陷入这样一种无谓的境地: 学术研究无法追求他们本应欢愉的大胆的, 异想天开的想法——仅仅因为他们受限于运行高度简化的模型.
当然, 我们可能会学习一些策略来减少有限资源的影响. 例如, 梯度检查点等技巧可能有助于减少内存占用. 另外, 局限性的存在很可能会促使我们创造性地设计其他模型, 能够以更低成本来实现类似或更好的性能——就像Baker小组对trRosetta所做的那样, 它以更小的架构超越了CASP13的AlphaFold. 然而, 很明显, 那些拥有更多计算能力的公司将始终占据上风.
这可能会导致未来计算研究小组需要在基础设施上进行大量投资才能生存——就像我们在实验生物科学领域的同事一样, 尽管设备的淘汰速度要快得多. AlphaFold的成功可能会让资助机构相信, 只要有足够的资源, 计算研究就能做大事, 并使之成为可能. 或者, 我们可能都得把资源集中起来, 组成一个大规模的国际财团, 大规模地购买硬件——就像高能物理学家们不得不联合起来建设欧洲核子研究中心这样的大型项目一样.
话题开始变得有点黯淡, 所以我打算以我们现在正在讨论研究经费为借口, 停止不经意的抱怨, 讨论另一个话题. 那就是几十年来, 主要由学术研究团体收集的大量蛋白质结构数据和信息所发挥的作用.
DeepMind成功的一个重要因素是技术的可用性, 尤其是结构生物学小组几十年来苦心收集的数据. 他们用于训练的蛋白质数据库(Protein Data Bank)收集了约17万个结构, 其中大部分是由学术团体制提供的, UniRef蛋白质数据库, 或宏基因组学序列的BFD和MGnify也是一样. 采用的软件工具, 如HHblits, JackHMMER和OpenMM也是由政府资助的学术计划开发的. 同样重要的是——这些计划中的大部分都是由公共资金资助的. 尽管DeepMind的战利品可能很大, 但纳税人大几个数量级的投资才使他们的成就得以实现.
这一点对由专业学者主导, 撰写和评审, 由同行评议的文章中关于蛋白质结构预测的大量研究同样成立 .这包括AlphaFold整合的许多想法, 从利用多序列比对来预测蛋白质结构, 到将模板纳入建模.这绝不是要贬低DeepMind的工作. 他们已经开发出了一种新颖的解决蛋白质结构预测的方法, 将许多创造性的想法与精湛的工程相结合. 但是, 如果说他们能看得更远, 那是因为他们站在巨人的肩膀上.
这就提出了许多关于研究伦理以及人工智能的有趣问题. 例如, 考虑一下Alphabet决定对AlphaFold进行商业开发的可能性——他们从这样一大批几乎完全由纳税人支付的研究中获利是否合理? 由可公开获得的研究创造的信息在多大程度上属于公众——请注意, 公开是为了刺激进一步的公共研究——以及在什么条件下它可以用于营利性的活动? 如果我们想让科学保持其应有的开放性和协作性, 就需要提出许多问题.
AlphaFold为什么强?
AlphaFold团队成员

虽然摘要中没有讲AlphaFold 2的具体细节, 先让我们来看看那19位共同一作都是做什么的吧!

第一位John Jumper是这届AlphaFold的领袖, 我记得上一届是Andrew Senior(此人CS背景). 这届明显换人了, 而且这位之前的研究经历是匹配这个问题的, 可以说是domain knowledge丰富. 他的linkedin公开简历如下
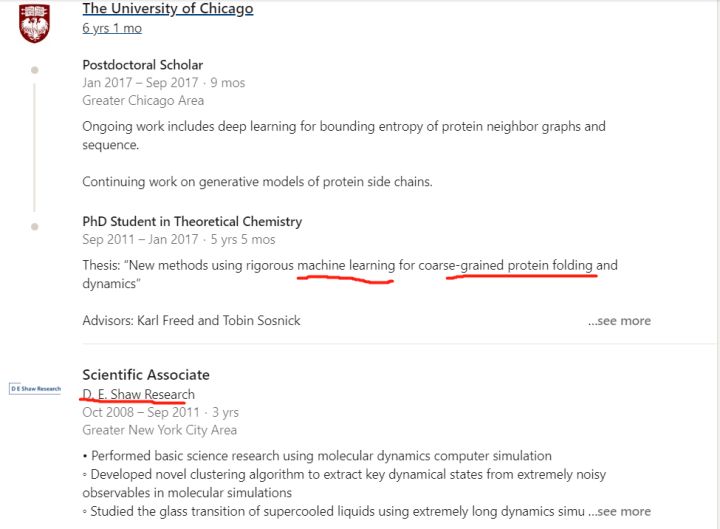
这位年轻帅气的大哥, 08-11年在世界上”最豪华”的分子动力学研究所, 也就是传奇的对冲基金大佬D.E. Shaw带领下的D.E. Shaw Research研究所的地方研究分子动力学模拟. 这人练级过程中有分子动力学和商业驱动科研的经历. 在芝加哥大学研究机器学习和粗粒化蛋白质折叠的方法. 最后进入了世界上又是”最豪华”的商业公司, 一个曾经把柯洁下棋下到流泪的公司, DeepMind, 开启了AlphaFold2的研究之路. 这人的履历就是为蛋白质折叠这个问题而生的!
关于此人的更多信息, 可以阅读其实验室同组人员王宗安的介绍AlphaFold 2领队Jumper奇人 - 我们需要什么样的复合型人材?.
第二位作者是Richard Evans, DeepMind有两个richard, 一开始把我搞蒙了, 找到一个AI方向, 具体细分是做范畴逻辑?(Cathoristic Logic)的, 原来的应用是, 个人行为和社会行为(Social Practices and Individual Personalities)? 据我脑子中的浅薄的知识, 目前所有学术界在做蛋白质结构预测方向好像都没有用上这个. 以为DeepMind在这里用了什么黑科技. 然后经提醒发现是这个Richard Evans, 主攻Deep reinforcement learning的, 且参加过初代AlphaFold的开发, 瞬间合理了!
第三位Alexander Pritzel, 理论高能物理背景, 最高引文章是还是deep reinforcement learning, 有理由相信AlphaFold在这个策略上进行了尝试.
第四位Tim Green, 量子化学, 凝聚态物理转移, 博士工作是利用密度泛函理论来预测NMR的耦合参数.
第五位Michael Figurnov, 相关工作有residual network, 这个在AlphaFold中已经部署.
第六位牛津大学博士Kathryn Tunyasuvunakool, 博士时候这位姐姐的主要工作在自述中应该是生物数据的处理, 她在博士期间还写过生物数据可视化的代码.
第七位Olaf Ronneberger, U-net的作者, 单篇被引用次数超过20000, U-net是用于生物医学图像分割的卷积网络.
第八位牛津大学博士Russ Bates, 医学图像处理.
第九位剑桥大学MEng, Augustin Žídek, 深度增强学习相关.
第十位Alex Bridgland, 牛津大学计算机博士, 资料不详, AlphaFold一代作者之一.
第十一位Clemens Meyer, 世界顶尖商学院巴黎高商毕业, 且有了十年以上管理经验的资深产品经理. 这个是我没有想到的. . .
第十二位Simon Kohl, KIT毕业物理硕士, CS博士, AlphaFold一代作者.
第十三位Anna Potapenko, 俄罗斯国立高等经济大学CS博士, 自然语言处理相关,
这篇文章在我能够理解的范围内, 瞎猜一个, transformer? long-range sequence?部署了transformer在提取序列attention当做input?(赛前猜的, 猜对了一半, 用了transformer直接end2end了)

第十四位Andrew Ballard, 计算物理学家, 2015年加入DeepMind, 用过副本交换, 研究过非平衡态系统, 可能可以用来解决后面蛋白质结构refinement的问题?
第十五位Angew Cowie, 参与开发了Acme!DRL!
第十六位Bernardino Romera Paredes UCL CS master.
第十七位Stanislav Nikolov, MIT MEng master.
第十八位, Rishub Jain, CMU CS master.
第十九位, Demis Hassabis 大boss.
这么多看下来, 这19位真是兵强马壮, John Jumper又是为这个方法而生的, 同时还引入了一个产品经理来维护整个团队, 这些都是学术界不大可能拥有的东西. 18年的AlphaFold我们还可以argue, 其实他是集学术界大成者, 整体创新性并没有那么强, 起码大家都很容易follow, 这次的AlphaFold 2我要下个暴论, 是真正意义上的应用了AI来解决蛋白质折叠问题!
计算资源
我之前对计算资源有点不屑一顾, 不就是128个TPU么, 几百万就搞定了, 能拿的出这钱的单位多了. 但是我现在想通一个问题, 就是算法的迭代. 研究人员在研究的时候要有无数次的尝试, 如果训练模型不能快速的给出反馈的话, 科研的进度就会被大大拖累. 有大量的计算资源, 不仅仅是提高了模型的复杂度而已, 而是提高了研发人员的速度. 就好比以前我们做gremlin开发的时候, 部署在matlab框架下要一天时间, 重新部署在tensorflow下几秒钟就够了, 于是我就可以尝试各种奇怪的idea了. 速度同样可以带来大量的创新.
所以我相信, 128个TPU只是最终模型训练的结果, 在研发过程中, 肯定调用了更多的你难以想象的计算资源!但是这也没有完, Baker团队的trrosetta用非常轻量的模型就超过了18年的AlphaFold, 所以在未来的几年, 对问题理解的更好, 学术界的平民版AlphaFold我相信也很快会出来的.
这对生物学意味着什么?
现在有两个重要的问题在大多数蛋白质生物信息学家中流传. 第一个问题是: 他们[DeepMind]会不会公开他们的代码, 如果会, 如何公开? 而第二个问题, 只是稍微不那么重要, 那就是: 运行它需要什么?
第一个问题是最重要的. 当被问及代码可用性的问题时(刚好超过虚拟[CASP14]问答聊天框中三分之一的问题), John Jumper声称他们正在DeepMind进行”内部讨论”, 关于”将他们的工作提供给社区”, 他们希望在1月初宣布.
可能出现多种情况. 从根本上讲Alphabet是一家私人的营利性公司, 所以他们可能会决定对AlphaFold 2进行商业化利用——就像OpenAI决定对GPT-3(今年早些时候推出的著名语言模型)所做的那样. 这也很可能意味着代码将保持私有性, 坦率地说, 这将至少在一段时间内阻碍蛋白质信息学的进展. 还有一种可能是, 他们决定将代码开源——可能是对商业用户的某种授权——这也是大家希望他们在论文通过同行评审后所做的事情, 这样社区就可以在这个令人难以置信的成功基础上再接再厉.
提供他们的代码并不意味着任何人都可以运行它. 当他们的Nature论文在去年1月发表时, 还缺少一个关键的部分: 构建神经网络的输入特征的代码. 虽然他们确实提供了这些特征的描述, 但我和一些OPIGlets尽管在这个方向上做了大量的努力, 却一直无法得到有意义的结果——从CASP14 Discord频道的讨论来看, 似乎许多其他科学家也做了类似的尝试, 结果同样令人失望. 然而, 有了这个架构, 应该可以完全重新训练这个模型, 或许可以通过汇集几个来源的资源, 部署一个虽然比AlphaFold 2稍差的系统, 但对于实际应用还是有用的.
这就引出了一个相关的问题. 我们知道DeepMind采用了大量的计算能力来制作AlphaFold 2, 但它实际运行的时间有多长? 当被问及训练和运行他们的模型需要多长时间时, John Jumper重复了DeepMind的博文信息, 即制作最终模型所使用的资源——但避而不谈运行代码需要多长时间, 以及在什么条件下运行. 新闻稿中提到的”几天”, 在128个TPU-v3核心中, 很可能意味着以一般计算组的资源计算几个月.
12月4日更新: Demis Hassabis(DeepMind的CEO)证实, 根据蛋白质的不同, 该模型需要在5-40个GPU上运行”几小时到几天”. 这不是很有参考价值, 因为DeepMind已经研究了相当广泛的长度和序列比对深度, 而且我们不知道他们使用的是哪种类型的GPU——例如, 如果是40个Titan GPU, 将是10万英镑以上的基础设施投资. 然而, 即使这个价格也比实验方法至少低一个数量级, 而且速度快很多. 事情看起来很有希望.
如果代码运行速度快, 那么它可以被加载到API中, 并被任何连接互联网的人使用, 就像GPT-3一样. 如果它需要特殊的硬件, 那么它可能会被限制在有能力维护一个高性能计算集群的计算小组中. 我个人的直觉是后者的可能性更大, 因为最初的AlphaFold在通用GPU上运行需要几天时间……而AlphaFold 2似乎比它的前辈大得多. 无论成本如何, 它很可能比实验性的蛋白质结构测定要快得多, 也便宜得多, 而后者往往需要数年和数百万美元.
药物发现的一个经典障碍——假设我们知道一个可靠的疾病靶点, 这是一个完全不同的故事——是缺乏可靠的晶体结构. 如果一个靶点的结构是已知的, 那么就有可能设计出一种能与活性位点最佳结合的化合物——这个过程被称为__基于结构的药物设计__——以及对所述分子进行工程设计, 使其具有溶解性和低毒性的特性, 从而使其成为有用的药物. 不幸的是, 有整个靶点家族——想想G蛋白偶联受体(GPCRs), 其成员是FDA批准的三分之一药物的靶点——其结构无法准确了解. 快速, 准确的蛋白质结构预测有望极大地提高药物研发的效率.
精确结构预测的另一个有趣的应用将是精确的蛋白质设计和工程. 有几个小组, 特别是David Baker的小组, 已经用类似的想法工作了一段时间. 然而, 这将取决于一些技术细节, 例如AlphaFold从浅层多序列比对中提取信息的能力, 我们将在下一节简单讨论.
总的来说, 廉价而准确的结构预测将是生物学的一个胜利. 蛋白质的功能依赖于结构——按需生成结构的能力有望让我们进一步了解生命.
这对结构生物信息学意味着什么?
这意味着我们可以专注于结构预测之外的其他问题.
因为这正是AlphaFold 2所解决的问题, 而不是像很多新闻稿所说的那样, 解决__蛋白质折叠问题__. DeepMind的代码不会提供一个多肽或多条链如何在几秒钟内组装成发挥作用所需的复杂结构的信息. 它只能提供晶体结构的准确估计, 这只是蛋白质构象的快照. 但蛋白质远不止如此——晶体结构并不一定能告诉我们全部的故事(看这篇论文的例子).
更重要的是, 虽然AlphaFold 2为蛋白质结构预测提供了一个 一般 的解决方案, 但这并不意味着它是 通用 的. CASP14的几个目标没有被成功预测, 说明有一些蛋白质家族需要进一步的工作. 当然, 这些目标并不能完全代表一个蛋白质组. 该模型是在蛋白质数据库上进行训练的, 众所周知, 蛋白质数据库偏向于容易结晶的蛋白质. 此外, 由于AlphaFold采用多序列比对作为输入, 它是否能处理浅层次或信息量不大的问题还有待观察, 例如在蛋白质设计这个非常重要的问题上, 在突变序列或有时在抗体序列中发生的例子.
折叠本身就是一个引人入胜的问题, 不仅对基础生物学有意义, 而且对生物医学也有意义, 因为它可以让我们更好地理解许多疾病, 蛋白质错误折叠或者作为病因, 或者引发可怕的后果. AlphaFold 2的成功可能会给我们提供一些启示, 如果我们能够分析神经网络是如何推断折叠结构的——但它也可能提供很少的知识, 因为解释的困难, 或者只是因为网络的推断不能很好地代表动态折叠过程.
蛋白质运动, 包括柔性和变构效应, 是蛋白质信息学的另一个明显的发展方向. 这些机制是蛋白质功能和传递信号的基本方式, 但可用来模拟这些现象的计算技术仍然非常有限. Dominik Schwarz, 我们的OPIGlets之一, 最近表明, 通过深度学习的距离预测编码了一些关于蛋白域柔性的信息. AlphaFold 2可能也能提供类似的见解.
另一个我非常感兴趣的领域, 是研究蛋白质与蛋白质之间的相互作用. 以抗体为例, 想一想: 在旁位(界面的抗体部分)和表位(抗原部分)之间有一组相互作用, 这是维持结合的基础. 尽管做了大量的工作, 蛋白质-蛋白质对接在很大范围内仍然不成功, 并且存在类似CASP的常规评估, CAPRI. 从AlphaFold 2中吸取的经验可能会刺激这个领域, 尽管我们从CASP14中知道, 它经常无法预测晶格接触.
最后, 蛋白质结构的可用性增加只会提高人们对蛋白质-配体对接的兴趣, 即预测配体将如何与蛋白质相互作用——以及作用有多么强烈. 尽管仍然普遍缺乏能够在这一领域取得重大进展的好的注释数据, 这一领域也已经取得了显著的进展, 特别是在新型深度学习方法相对成功的情况下.
学术问题
蛋白质折叠问题解决了没有?结构基因组学时代来临了没有?在这个技术下面, 哪些目前的技术会被替代?结构生物学的空间在哪里?哪些有瓶颈的技术会得到突破, 比如和蛋白质组学联合解释数据?
基本解决了, 从结构生物学的角度讲, 基因平等, 然而人类总是挑一些可能比较有意思的蛋白去解析结构, AlphaFold 2预测的精度足够高, 一些犄角旮旯的蛋白结构可以得到大量的补充. 而且在序列数据爆炸的情况下, 可以得到大量可靠的预测模型是非常有意义的. 按Nature的一篇评论, 人们可以花更多的时间思考, 花更少的时间拿移液枪了.
但是AlphaFold也提了, 氨基酸侧链的精确位置仍然是一个挑战, 还有一些比如PPI, DNA, RNA, 小分子配体的结合还没有解决. 制药行业对侧链的精确度是非常之高的.
机制问题
学术界干了几十年没解决的蛋白质折叠问题, DeepMind为什么做的这么好, 仅仅是因为资源丰富吗?
除了算法强之外, AlphaFold还汇集了几个领域的大佬, 甚至请了一个专业的产品经理, 学术界的合作是否能如此的紧密?目前的学术运营框架下合作难度多大?
学术界是不是在搞跳高运动, 每年创新1cm?
社会问题
对学术界有什么影响?
I think it’s fair to say this will be very disruptive to the protein-structure-prediction field. I suspect many will leave the field as the core problem has arguably been solved,” he says. “It’s a breakthrough of the first order, certainly one of the most significant scientific results of my lifetime.”
Mohammed AlQuraishi
网红科学家穆罕穆德, 这对这个领域是破坏性的, 这个领域的核心问题已经被解决了, 我想很多人都会离开这个领域了吧.
结论
如果不是从认识论的角度, 而是从影响的角度来看的话, 谷歌刚刚取得的成就很可能是本世纪最重要的科学成就之一. 长期吃香的从蛋白质的序列预测蛋白质结构的能力(以及类似的突变序列的可得性)将开启从基础生物学到医药应用等整个生命和医学科学的应用. 前景确实令人震惊.
话虽如此, 但这句话还是要慎重对待. 虽然我们已经有了解决蛋白质结构预测问题的 一般 方案, 但还没有一个 通用 的方案. CASP中的一些结构即使AlphaFold 2预测的准确性也很低, 这表明对特定的目标家族可能需要做进一步的工作. 众所周知, 用于训练的蛋白质数据库偏向于易结晶蛋白质, 目前还不清楚这将如何影响其对暗蛋白质组的作用. 此外, 由于预测依赖于多序列比对, 当比对中的序列很少或没有序列时, 如设计蛋白, 或者当它的信息不充分时, 如抗体, 这种方法是否有效还有待观察.
DeepMind的成功也提出了一些科学界需要相当认真考虑的问题. 虽然他们比大多数个人研究小组更灵活, 资金也更充足, 但这一成就却引出了一些深层次的问题, 那就是我们进行研究和交流的方式, 以及我们这个拥有更多资源和积累知识的群体是否真的有效利用了它们的潜力. 我们还需要反思我们作为科学家的责任, 确保科学保持开放, 确保在公众支持下进行的研究对公众仍然有用.
抛开这些顾虑不谈, 结构预测问题的解决将最终刺激新的研究路径. 长期以来, 我们一直专注于再现通过X射线晶体学捕捉到的蛋白质结构的静态图景. 现在我们可以把更多的精力投入到其他同样有趣的问题上: 蛋白质是如何折叠成这些奇异复杂的构象的? 它们是如何运动的, 这种运动又是如何调节的? 它们如何与其他蛋白质以及配体相互作用? 这只是蛋白质信息学一个非常令人兴奋的时代的开始.
我要感谢Mark Chonofsky, Fergus Imrie, Constantin Schneider, Javier Pardo Díaz, Matthew Raybould和Garrett M. Morris, 他们辛勤地审阅, 识别这篇文章的错别字, 并为这篇文章的第一稿和第二稿提供了宝贵的反馈意见.
更多讨论
- 如何看待 AlphaFold 在蛋白质结构预测领域的成功?
-
[专访 AlphaFold2是一艘曲率飞船](http://blog.sciencenet.cn/blog-3458695-1261414.html) - AlphaFold2 解决了蛋白质结构问题吗?DeepMind 解决这项生物学五十年难题有何重大意义?
