- 2021-06-13 00:34:41 整理: daiyx; 校订: 李继存
VMD自带了一个氢键分析插件hbonds, 可以基于VMD内置功能分析轨迹中氢键的存在数目以及占据率(也可以称为频率), 但所用的判断标准与GROMACS的存在区别, 具体的细节可以参考GROMACS和VMD中的氢键判定标准.
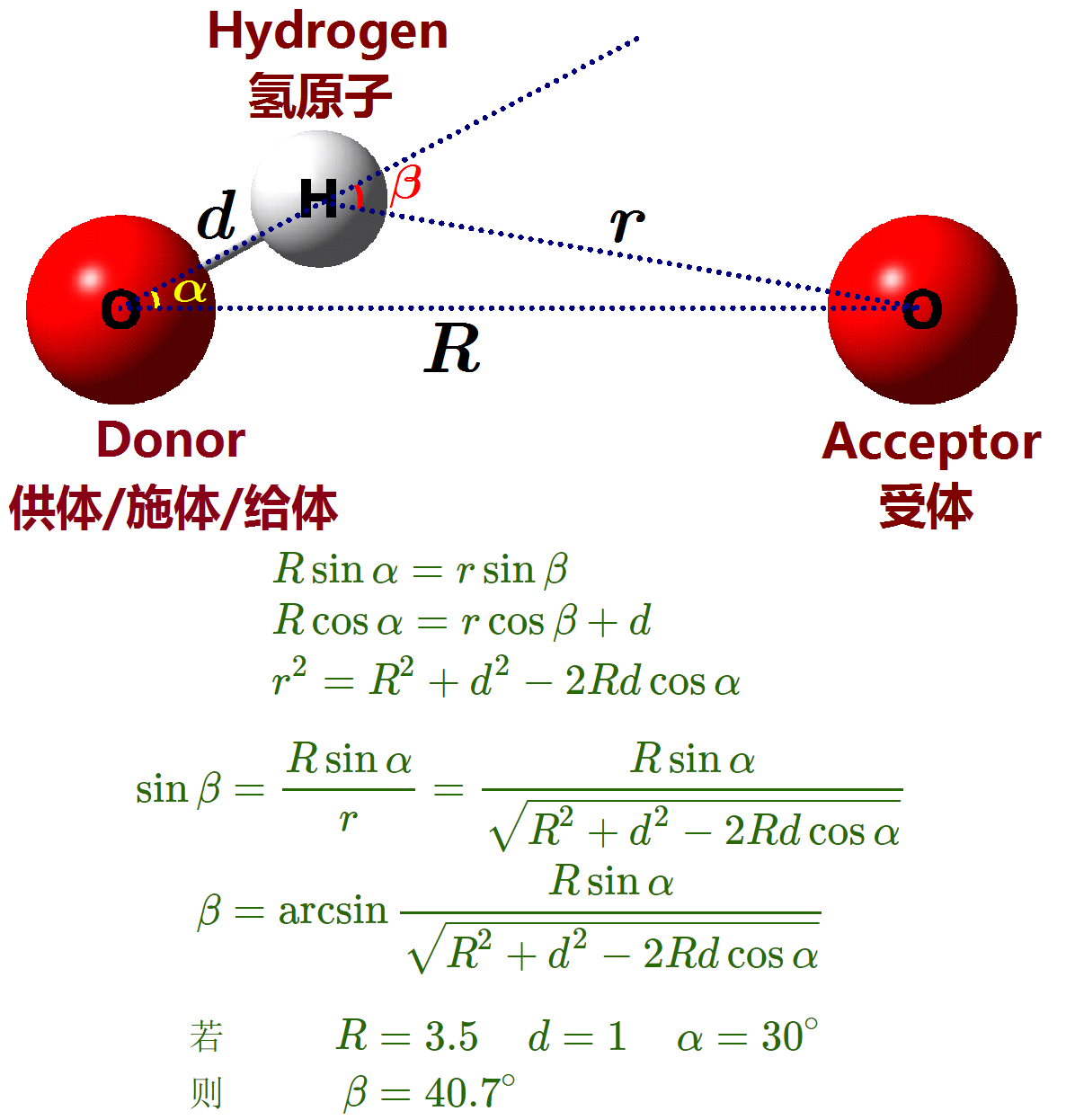
简而言之, GROMACS默认用的 $R-α(3.5-30)$ 标准, VMD则用的 $R-β(3.0-20)$ 标准. 没法精确地在两个标准之间转换, 如果VMD要使用GROMACS的标准, 近似的设置为 $R-β(3.5-40)$.
此外VMD在计算氢键时不会考虑周期性边界条件, 所以对于满盒子的体系, 所得结果存在问题.
hbonds 1.2插件说明文档
hbonds插件主要用于计算整条轨迹中形成的氢键数目。搜索氢键时可以在单个选择或两个不同的选择之间进行,用户也可以指定帧的范围。
形成氢键的标准
如果供体原子D(或称给体, 施体)与受体原子A之间的距离(即D-A距离)小于距离截断值(默认为3.0 Å),并且D-H-A角度小于角度截断值(默认为20°),则认为在结合氢的原子(供体D)和另一个原子(受体A)之间形成氢键。
选项
插件提供的选项包括, 最多两个选择(原子不应该有重叠), 要计算的帧。每帧都可以更新选择,但代价是速度慢, 关于选择时可能需要此选项的详细信息,参见Salt Bridges插件。插件可以马上在 VMD 内绘制氢键数目随时间变化的图像,也可以保存到文件(默认为hbonds.dat)。此外,所有消息都可以输出到一个日志文件。
插件提供了进一步的选项来控制选择中使用的原子(只考虑极性原子, 还是考虑VMD通常使用的所有原子),是否限制第一个选择是供体、受体或两者兼有(默认为两者兼有),以及是否计算有关氢键的详细信息。详细的输出包括在轨迹中形成的所有氢键(根据一些基本的标准)及其频率。请注意,当使用选项all详细输出时,相互作用频率可能大于100%,因为给定残基对可能包含多个氢键,每个都会单独计数。
命令行接口
该插件的所有功能都可以通过命令行接口获得.
用法:hbonds -sel1 <atom selection> <option1> <option2> ...
选项:
-sel2 <原子选择>(默认:none)-writefile <yes|no>(默认:no)-upsel <yes|no>(每帧更新原子选择. 默认:yes)-frames <begin:end> 或 <begin:step:end> 或 all 或 now(默认:all)-dist <供体和受体之间的距离截断值>(默认:3.0)-ang <角度截断值>(默认:20)-plot <yes|no>(使用MultiPlot绘图, 默认:yes)-outdir <输出目录>(默认: 当前目录)-log <日志文件名称>(默认: 无)-writefile <yes|no>(默认:no)-outfile <dat文件名称>(默认:hbonds.dat)-polar <yes|no>(只考虑极性原子, 即N, O, S, F? 默认:no)-DA <D|A|both>(sel1视为供体(D), 受体(A), 还是既可视为供体也可视为受体(both)). 只有使用两个选择时此选项才是有效的. 默认:both)-type: (默认:none)none不计算详细的成键信息all相同残基对类型中的氢键进行完全计数pair相同残基对类型中的氢键只计数一次unique根据供体-受体原子对类型计数氢键
-detailout <详情输出文件>(默认:stdout)
作者
- JC Gumbart
- Dong Luo (
us917@yahoo.com)
应用: 分析显示两个分子之间氢键
简单的使用没太多可说的, 只要弄明白每个选项的含义即可.
下面考虑一个复杂点的应用: 使用VMD只显示两个分子间所成的氢键, 即不能显示每个分子内的氢键.
显示氢键可以使用VMD的氢键显示模式, 但这种显示模式会显示所有的氢键, 而无法只显示不同分子之间的氢键. 但有时候在分析时为了便于查看, 我们希望能够只显示分子之间的氢键, 而不显示分子内部的氢键. 比如, 对于蛋白-配体复合物, 如果用VMD的氢键模式进行显示, 会同时显示出来蛋白-蛋白之间的氢键, 配体-配体之间的氢键, 蛋白-配体之间的氢键, 而我们只需要显示最后一项. 在对一些包合物进行分析时, 存在同样的问题.
为此, 我们需要做的是, 获得所有可能参与形成分子间氢键的原子, 将对其应用氢键显示模式. 如果分子间氢键所涉及的原子数目不多, 手动获取即可, 否则的话, 我们可以借助hbonds插件获得. 但这个插件默认不会输出涉及形成氢键原子的编号, 所以我们需要对其源代码进行简单的修改, 让其输出我们需要的原子编号.
修改hbonds插件源代码输出原子编号
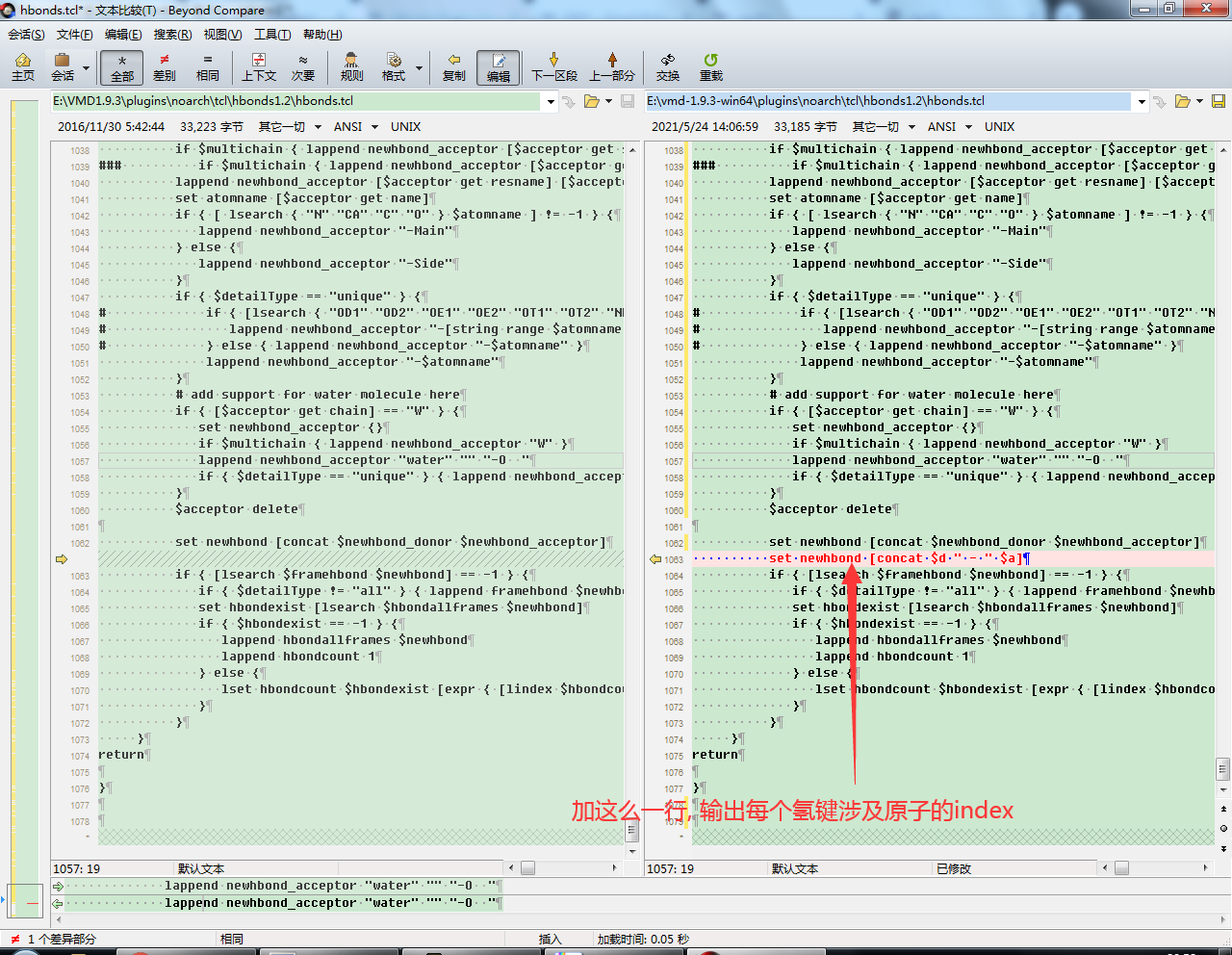
hbonds插件的源代码位于【VMD安装目录】\plugins\noarch\tcl\hbonds1.2\hbonds.tcl. 代码写的有点乱, 但不难理解. 对于我们的目的而言, 最小的改动就是在1062行后添加一行set newhbond [concat $d "-" $a],将输出改为每个氢键涉及的氧原子,然后保存。
获取轨迹的氢键原子编号
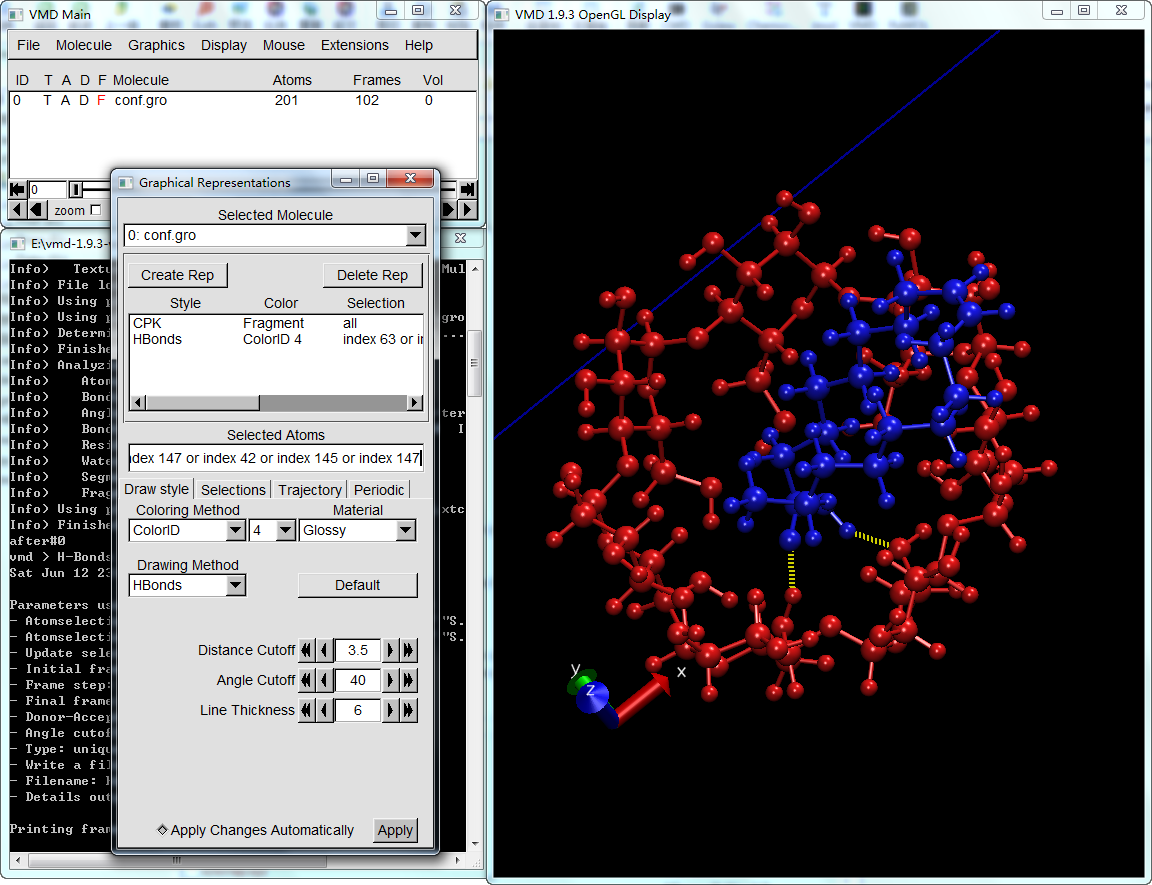
以GROMACS包合物轨迹为例. 加载gro文件和xtc轨迹, 点击VMD Main窗口Extensions工具栏下Analysis中的Hydrogen Bonds, 打开氢键插件,照下设置计算参数, 注意保存输出文件。
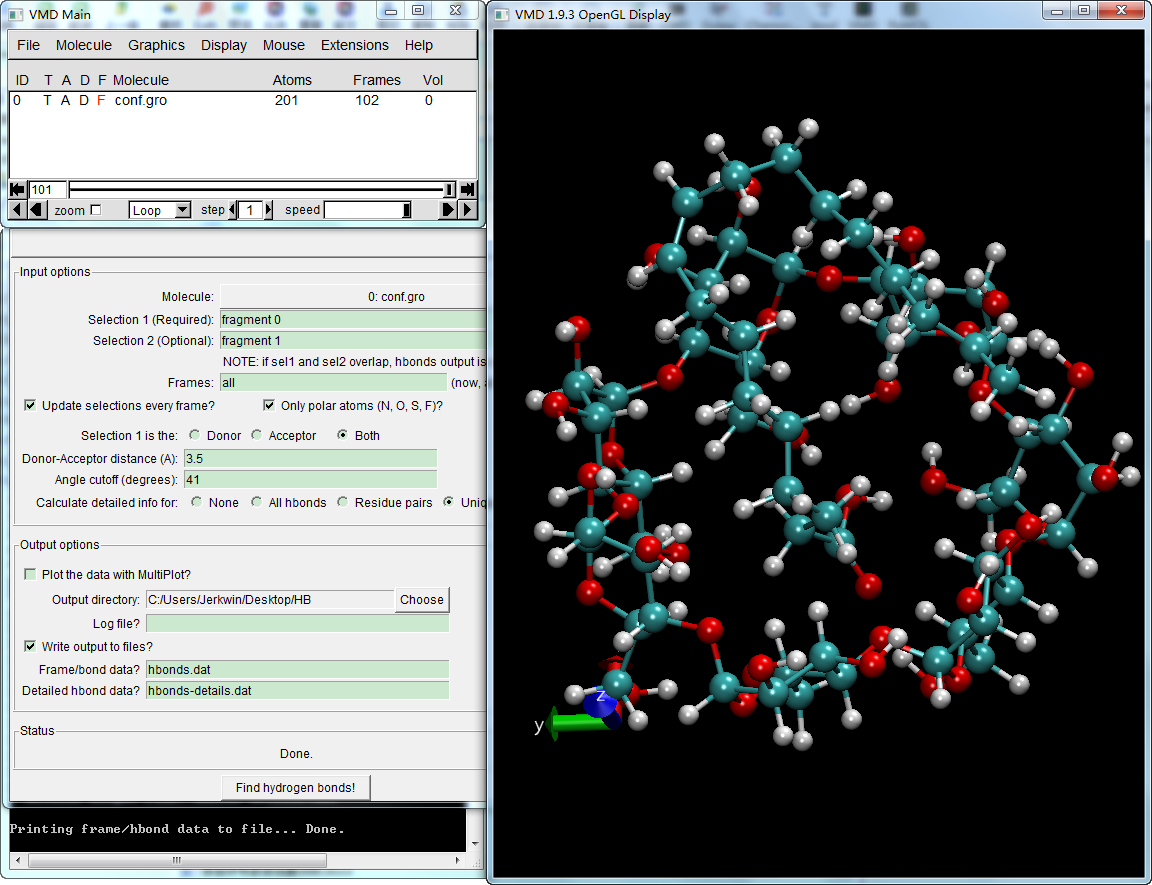
设置好后, 点击最下面的Find hydrogen bonds!, 等待完成后, 即会在工作目录下生成两个文件:
hbonds.dat: 每帧的氢键数目, 第一列为帧号, 第二列为氢键数目.hbonds-details.dat: 所有氢键的供体, 受体, 占有率. 内容类似如下
Found 3 hbonds.
donor acceptor occupancy
63-148 100.00%
147-42 84.31%
145-147 69.61%这样我们得到了可能氢键的总数及涉及的原子编号。
将氢键原子编号整理为选择语句
将前一步得到的hbonds-details.dat中的氢键原子编号整理为选择语句, 类似index 63 or index 148 or index 147 or index 42 or index 145 or index 147, 这就是所有可能参与分子间氢键形成的氧原子的编号. 重复的编号不会导致问题, 虽然如果涉及原子数过多时可能导致选择语句多长. 不确定VMD的选择语句最多支持多少字符. 如果过长的话, 也可以将其拆分为多条.
使用氢键模式显示选择的原子
以前一步的选择语句创建新的rep, 然后以HBonds模式显示, 同时调整判断氢键的标准与前面插件使用的相同, 这样就可以显示每帧的分子间氢键了。
这是直接以氢键模式显示所有原子的结果, 可以看到分子内的氢键也会显示出来
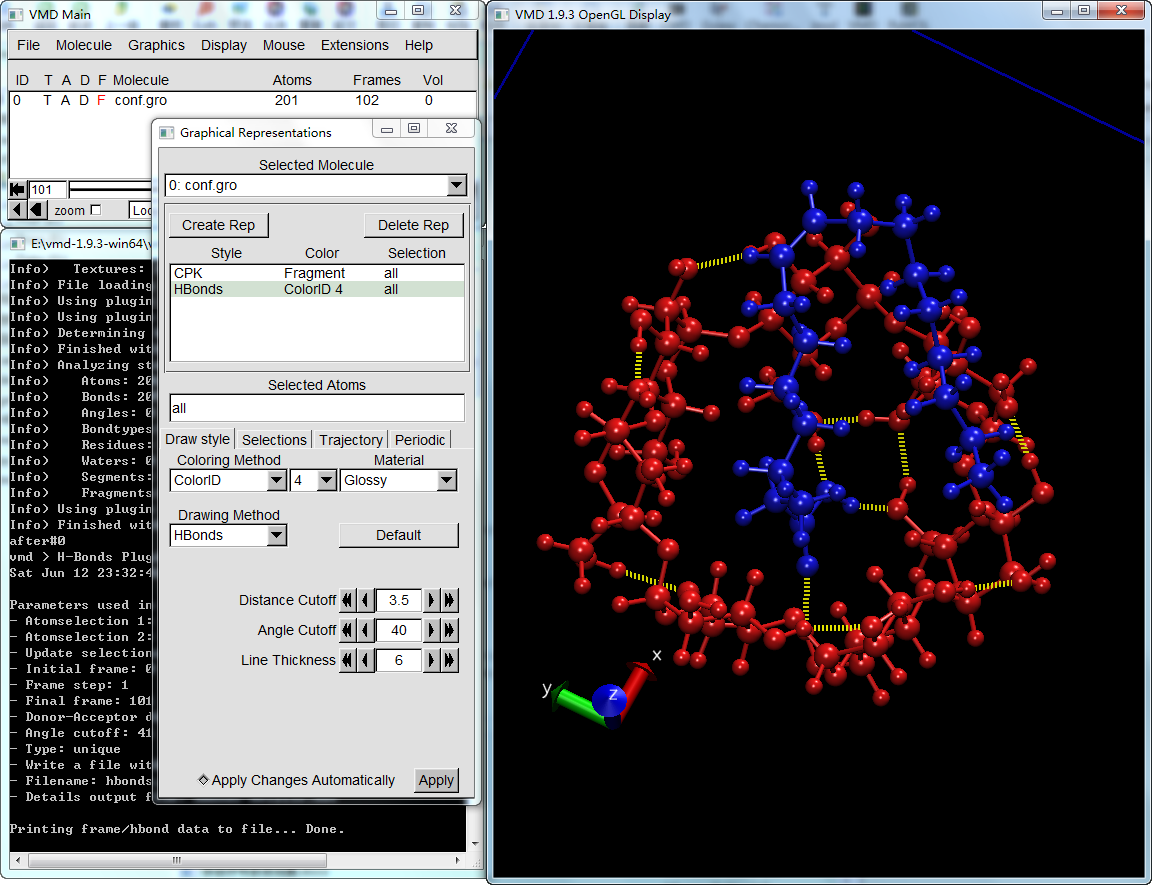
这是只将氢键显示模式用于选择原子的结果, 只显示了分子间的氢键.
播放轨迹, 看看对每帧是否正确显示. 也可以与前面得到的hbonds.dat中的数目进行对照.
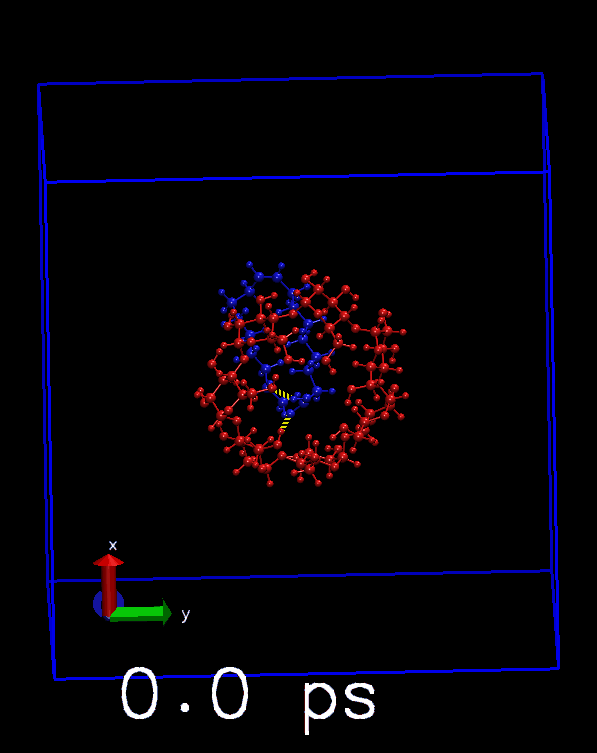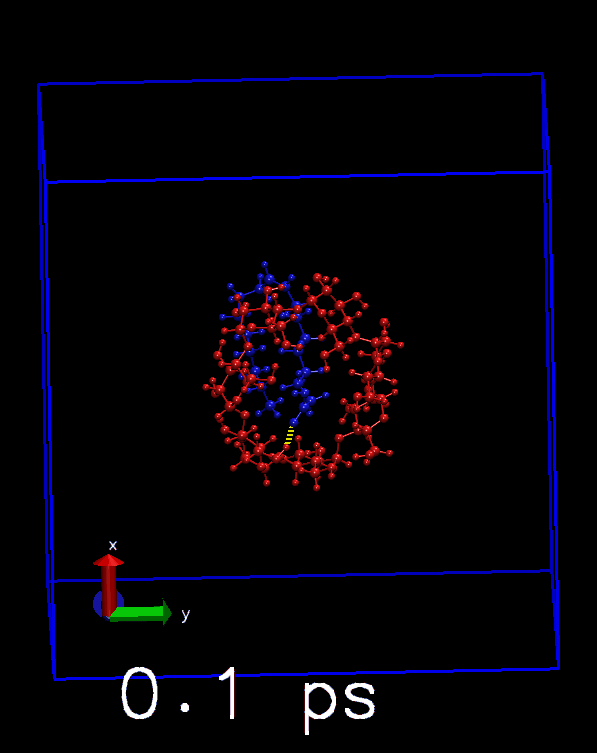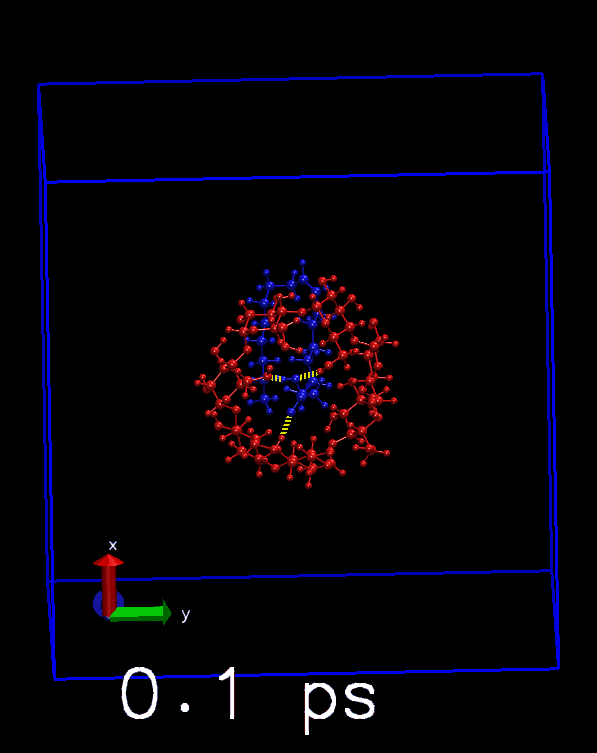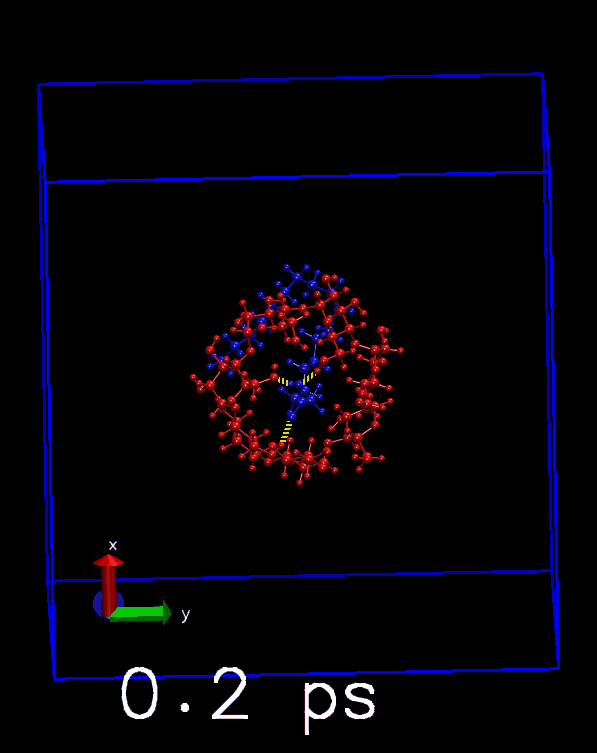
其他
- 对于PBC的问题, 除了直接修改VMD源代码外, 也可以自写tcl脚本来处理.
- 对于不同氢键判断标准问题, 处理方法同上.
- 自己写tcl脚本绘制氢键, 也没有太大问题, 具体方法可参考Pi-Pi堆积距离和堆积角度的计算.
