- 2022-08-20 00:09:10
VMD说到底是一个三维图形显示程序, 只不过主要用于显示较为简单的分子结构, 不太涉及过于复杂的三维模型. 背后的图形显示方式与一般的三维建模或渲染程序类似, 但也有些区别. 我们这里较细节地考察一下, 给定一组原子坐标, VMD到底是如何将其展示在屏幕上的. 这其中涉及很多的矩阵变换, 属于计算机图形学的内容, 就当作对计算机图形学的学习吧.
计算机图形学中常会用到几个不同的空间, 也就是不同的坐标系:
- 模型空间M, 也称对象空间, 局部空间: 模型自身所在的空间, 也就是创建模型时所使用的坐标系, 常取为主轴系. 对分子而言, 就是用于定义分子坐标的那个坐标系.
- 世界空间W, 也称全局空间: 场景中的所有对象, 如模型, 光源, 相机等所处的空间. 通常用于表示要渲染对象之间的空间关系. W比M更高一级, 模型可以在W空间中平移, 旋转, 缩放, 因此, 其W空间中的坐标值一般情况下会与其M空间中的值不同.
- 相机空间C, 也称观察空间: 跟W类似, 但其原点为相机或观察者, 一般取相机(视线)方向为Z轴. 屏幕上最终显示的图形是通过某个相机所观察到的, 相机观察时所用的空间就是相机空间, 它取决于相机在世界空间中的位置和取向.
- 投影空间P: 用来将C中的对象进行投影变换, 并将X, Y可见范围限制在[-1,1],Z限制在[0,1].
- 屏幕空间S: 前面几个空间都是三维的, 但最终显示的图形是二维的, 所以需要将P中的三维坐标缩放至二维. 在这个二维空间中, 一般左上角表示屏幕坐标原点, X水平向右, Y轴垂直向下, 宽w像素,高h像素.
不同空间之间的转换涉及很多坐标变换, 大多是3三维, 四维矩阵的操作. 类似如下
M–世界(模型)变换–>W–观察(相机)变换–>C–投影变换–>P–视口变换–>S
如果这些空间中有些是重合的, 也就是二者的坐标系原点, 取向都相同, 那么就可以省略变换中的一部分, 因为涉及到矩阵是单位矩阵. 有时候, 我们会将世界空间, 相机空间, 屏幕空间重合, 合称视图V空间, 这样最终只需要三个空间, M, V, P, 相应的矩阵是计算机图形学中三个最常见, 最常用的矩阵. 有时甚至会将M, V也重合, 这样就可以直接将模型投影到屏幕了.
在这么多空间中穿梭变换, 有时很容易混淆迷惑, 咫尺天涯, 不知此身何处. 这种时候, 建议重新归位到世界空间. 可以将其视为静止的绝对空间, 虽然可能不会明确用到它, 但可以将其作为其他所有空间的背景, 就像物理学中的绝对时空背景一样. 这种做法类似物理研究的方式, 先定义下一个世界空间, 将模型, 相机等放置到世界中, 然后在世界空间中进行操作, 虽然有时采用特定的空间可能更方便.
测试
我们测试一个简单的分子模型
| mol.xyz | |
|---|---|
1 2 3 4 5 6 | 4
mol
C 0 0 0
O 1 0 0
Cl 0 1 0
N 0 0 1 |
将其载入VMD, VMD默认显示如下
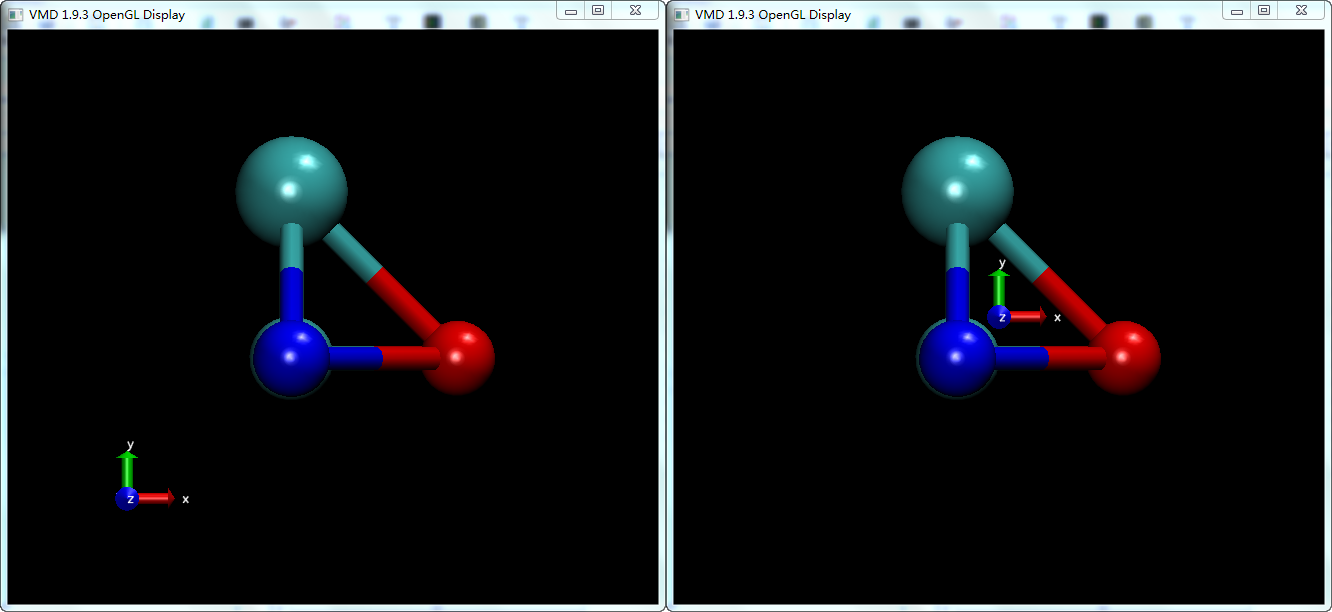
左边: 设置坐标轴处于左下方; 右边: 设置坐标轴处于原点
下面我们使用tcl脚本获取VMD使用的几个矩阵
中心矩阵center_matrix
> set C [molinfo top get center_matrix]
{ {1 0 0 -0.25} {0 1 0 -0.25} {0 0 1 -0.25} {0 0 0 1}}这代表分子模型空间的原点相对于世界空间原点的偏移, 由于世界空间的原点为分子的几何中心, 所以也就是分子模型空间原点相对于其几何中心的偏移.
旋转矩阵rotate_matrix
> set R [molinfo top get rotate_matrix]
{ {1 0 0 0} {0 1 0 0} {0 0 1 0} {0 0 0 1}}为单位矩阵, 因为我们尚未进行任何旋转操作. 如果使用鼠标或rotate命令对分子进行了旋转, rotate_matrix矩阵就会改变.
缩放矩阵scale_matrix
> set S [molinfo top get scale_matrix]
{ {1.5 0 0 0} {0 1.5 0 0} {0 0 1.5 0} {0 0 0 1}}这是默认的缩放大小, 具体值取决于分子尺寸, VMD默认会采用适当的缩放系数以保证整个分子都可见. 如果使用鼠标滚轮进行了缩放, 相应的矩阵就会变化.
全局矩阵global_matrix
> set G [molinfo top get global_matrix]
{ {1 0 0 0} {0 1 0 0} {0 0 1 0} {0 0 0 1}}这也是单位矩阵, 因为我们尚未进行任何平移. 如果进行了平移, 全局矩阵就会变化. 注意全局矩阵中只有平移部分起作用, 其旋转部分无法通过鼠标或菜单改变.
应用
通过上面的测试, 我们可以推断出, VMD的图形显示方法较为简单, 给定一组原子坐标后, 世界坐标系的原点取为这组原子坐标的几何中心, 世界坐标系的取向与原子模型坐标系的取向一致, 而相机坐标系和世界坐标系重合, X, Y轴处于屏幕内, Z轴垂直于屏幕向外. 大部分分子显示程序默认都是这种设置.
理解了图形显示的原理和VMD的实现, 我们就可以操控相关矩阵以改变视图. 这种操作不会改变分子的原始坐标, 只是改变了显示, 相当于相机在动. 由于相机相对于分子是固定的, 所以也可以认为是分子的模型空间在变而相机不变, 这两者的效果是一致的.
下面我们通过视图调整来实现几个简单的功能, 以示例这些矩阵的使用.
恢复默认视图
对分子进行了各种旋转平移缩放后, 恢复载入时的默认显示方式. 实现这个功能的菜单或命令为display resetview.
实现时只要将所有矩阵置为单位矩阵即可.
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 | set homeView {
{ {1 0 0 0} {0 1 0 0} {0 0 1 0} {0 0 0 1}}
{ {1 0 0 0} {0 1 0 0} {0 0 1 0} {0 0 0 1}}
{ {1 0 0 0} {0 1 0 0} {0 0 1 0} {0 0 0 1}}
}
proc homeview {} {
global homeView
molinfo top set {center_matrix rotate_matrix scale_matrix} transidentity
} |
保存当前视图, 并在需要时重新载入
VMD有几个插件可以实现这个功能, 且功能更多, 这里只是示例如何做.
| tcl | |
|---|---|
1 2 3 4 5 6 7 8 9 10 | set currentView $homeView
proc dumpview {} {
global currentView
set currentView [molinfo top get {center_matrix rotate_matrix scale_matrix}]
puts $currentView
}
proc loadview {} {
global currentView
molinfo top set {center_matrix rotate_matrix scale_matrix} $currentView
} |
根据当前视图, 旋转分子, 使其新坐标的默认视图与当前视图一致
前面说过, 改变视图并不会改变分子的坐标, 只是相机在动. 但有时候, 我们调整好了视图后, 希望以后再打开时VMD默认显示调整好的视图. 这可以通过保存VMD的状态文件来实现, 但稍微繁琐些, 因为我们不想另外保存一个与分子坐标文件配套的状态文件. 为此, 我们可以将分子的坐标进行变换, 使其载入后的默认视图就是我们需要的视图, 这样只要打开分子坐标文件, 不进行任何操作, 就可以得到我们需要的显示方式.
做法也比较简单, 根据前面所说的显示原理, 我们需要对分子坐标使用的旋转矩阵就是rotate_matrix.
| tcl | |
|---|---|
1 2 3 4 | set R [molinfo top get rotate_matrix]
set topmol [atomselect top all]
$topmol move [lindex $R 0]; # 注意这里的写法, 由于vmd设计的失误, 必须这样写
$topmol writepdb "mol~rot.pdb" |
我们也可以对轨迹中的每一帧进行这种操作, 具体代码就作为思考题吧.
直接修改矩阵
只是用来示例如何直接修改几个矩阵的值, 这样有需要的时候可以不用借助于VMD提供的那些矩阵操作来获得矩阵, 虽然可能麻烦点, 但不受限制.
| tcl | |
|---|---|
1 2 3 4 5 6 | set C [molinfo top get center_matrix]
lset C {0 0 3} -1
lset C {0 1 3} -1
lset C {0 2 3} -1
molinfo top set center_matrix $C
puts [lindex [lindex [lindex $C 0] 0] 3] |
